
Abstract
Muscular weakness and hypotonia may be associated with multisystem involvement giving rise to complex phenotypes, many of which are uncharacterized. We report a patient presenting with congenital hypotonia and severe ocular and brain abnormalities, evoking a Muscle Eye Brain disease (MEB). She had global muscular weakness, hypotonia and amyotrophy, joint hyperlaxity, kyphoscoliosis, respiratory insufficiency, dysmorphic features and severe intellectual disability. Brain MRI showed cortical atrophy and hypoplasia of the corpus callosum. Normal CK levels, non-progressive course and absence of dystrophic features or α-dystroglycan abnormalities on the muscle biopsy were not typical of MEB. CGH array identified a large de novo duplication in chromosome 11, including regions partially duplicated in three other patients with common clinical features. This report adds to the differential diagnosis of complex phenotypes characterized by muscular, ocular and CNS involvement and highlights the potential contribution of still unrecognized chromosomal abnormalities to these phenotypes.
Keywords
INTRODUCTION
Primary muscle diseases can be associated with multi-organ involvement. This is the case for dystroglycanopathies, a group of heterogeneous disorders characterized by absent or reduced functional glycosylation of α-dystroglycan due to mutations in one of 18 known causative genes [1]. Dystroglycanopathies are associated with a broad range of clinical phenotypes, ranging from severe congenital forms such as Walker Warburg syndrome or Muscle Eye Brain (MEB) disease, with structural ocular, brain and muscle abnormalities, to milder variants of limb girdle muscular dystrophy with no evidence of central nervous system involvement [2–6]. Mitochondrial myopathies can also present with a wide range of clinical signs and symptoms, typically with multisystem involvement [7]. Conversely, myopathic nonspecific features can be found together with other congenital abnormalities in different syndromic entities, many of which are unrecognized or not fully characterized yet. Borderline phenotypes are common, and raise differential diagnosischallenges.
On the other hand, chromosomal microarray techniques are increasingly used for genetic diagnosis of patients with unexplained developmental delay or multiple congenital abnormalities. In this respect, copy number variations (CNVs) have been identified in more than 30% of the human genome [8–10]. Although they have been pointed out as one of the most common causes of human disease, their role in muscle diseases is poorly understood.
Here, we report a patient with a congenital phenotype characterized by ophthalmologic, CNS and muscular involvement associated to an unreportedde novo duplication in chromosome 11 and discuss its implication for the differential diagnosis of congenital muscle disease.
CASE REPORT
The patient was the second female child born to healthy non-consanguineous parents of Mexican origin. Her sister is asymptomatic and there was no relevant family history. She was born at 38 weeks by cesarean section after an uncomplicated pregnancy. Marked hypotonia was noted since birth. At 2 months of age, a congenital hip dislocation was diagnosed and surgically corrected. Shortly after, her parents became concerned about delayed motor milestones and psychomotor retardation. At age 6 months, she was operated for a congenital bilateral closed-angle glaucoma. At 2.5 years, she had a rhegmatogenous retinal and choroidal detachment on the left eye. Surgical intervention was unsuccessful resulting in a subsequent left eye amaurosis. Shortly after, trabeculectomy and a valve implant for glaucoma control were performed on the right eye, in spite of which she experienced rapidly progressive vision loss. She also suffered from recurrent respiratory infections and severe gastroesophagealreflux.
When first evaluated at a muscle clinic at age 2.5 years, neurological examination showed global hypotonia and hyperlaxity, generalized and severe amyotrophy with proximal weakness, no joint or spinal contractures, bilateral hip subluxation, genu recurvatum, flat feet with hammer toes and pectus excavatum (Fig. 1). She had dysmorphic facial features such as synophrys, broad nasal root, hirsutism and low-set frontal hairline. Reflexes were abolished throughout and she had bilateral flexor cutaneous-plantar reflex. She was amaurotic in the left eye and had a multidirectional nystagmus in the right eye with a severely reduced visual acuity. She was able to sit upright unsupported but she got up from supine to sitting by rolling over and pushing on her arms. She had speech delay and communicated with gestures and facial expressions.
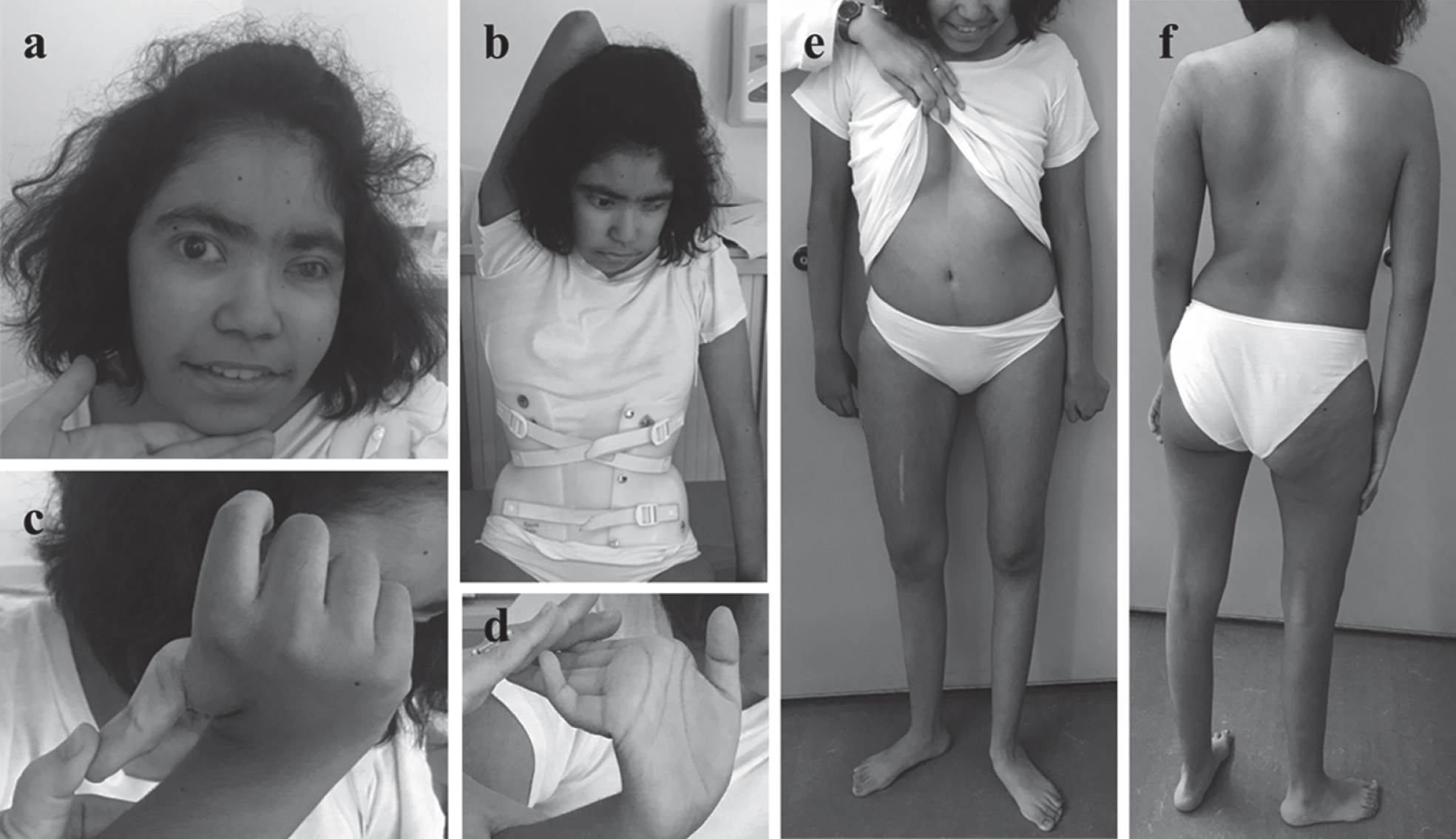
Clinical features at last examination (age 16 years). Global hypotonia and muscular weakness remained stable. Amyotrophy affected mainly the shoulder girdle and lower limbs (e,f). She had severe progressive kyphoscoliosis improved by bracing (b). Note dysmorphic facial features (i.e broad nasal root, synophrys, hirsutism, low anterior hairline) (a), thoracic deformities (pectus excavatum) (e), and prominent joint hyperlaxity (b,c,d) including flat feet (e,f). Permission for the publication of photographs was granted by the patient’s legalguardians.
CPK levels were normal (103 U/L). Sensory and motor nerve conduction studies performed at age 1.3 years disclosed no abnormalities and electromyography showed a myopathic pattern with decreased insertional activity, early recruitment pattern and short, small, polyphasic motor unit action potentials. Brain MRI at the age of 2 years showed diffuse cerebral cortical atrophy as well as hypoplasia of the Corpus Callosum (Fig. 2).
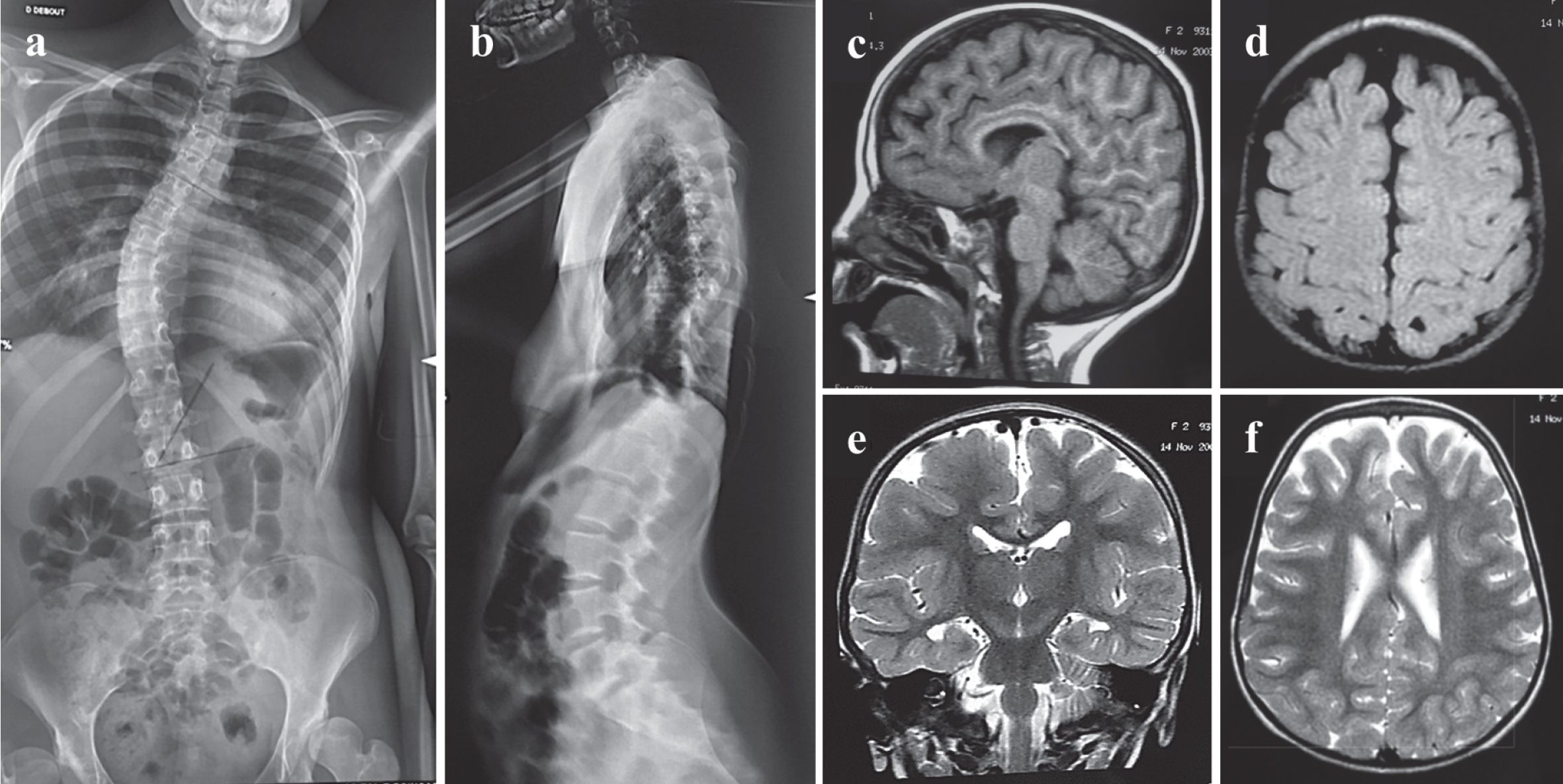
Radiological findings. Spine X-ray images (AP (a) and lateral projections (b)) at age 15 years showing severe kyphoscoliosis (44° Cobb angle) and lumbosacral hyperlordosis. Cranial MRI at age 2 years: sagittal T1-weighted image (c) reveals hypoplasia of the corpus callosum with prominent involvement of the splenium segment. Axial T1-weighted image (d), coronal (e) and axial T2-weighted (f) images show cerebral atrophy with prominent cortical sulci mainly in temporal, frontal and parietal lobes.
A quadriceps muscle biopsy obtained at age 1.5 years was reported to show nonspecific myopathic features (i.e. fiber size variation and scattered fibers undergoing necrosis and regeneration) and normal dystrophin immunostaining. Electron microscopy analysis was unremarkable. At age 2.5 years, a deltoid muscle biopsy was performed (Fig. 3), showing mild variation in fiber size and type 2 fiber atrophy. Immunostaining for dystrophin, α- and β- dystroglycans (VIA4-1- α-DG and NCL- β-DG, done twice), α-sarcoglycan, dysferlin, merosin-laminin α2 (80 kDa, 300 kDa and 4H280), laminin β1 and γ1 chains, collagen VI and perlecan disclosed no abnormalities. Dermal fibroblast culture with collagen VI immunolabeling found no abnormalities of collagen production and secretion. A Next generation sequencing (NGS)-based gene panel excluded mutations in 18 genes associated with dystroglycanopathies (Supplementary Table 1). Analysis of the muscle respiratory chain enzymes revealed raised citrate synthase and SDH activities, suggestive of potential mitochondrial overload or proliferation with no clear enzyme activity deficiency. Eventually, Southern blot and qRT-PCR analysis of mitochondrial DNA and POLG gene sequencing showed no abnormalities. Plasma lactic acid, coenzyme Q10, liver function, thyroid panel blood tests and plasma amino acids were normal. Normal urinary electrolyte levels and urine organic acids determined by liquid chromatography ruled out a Lowe syndrome.
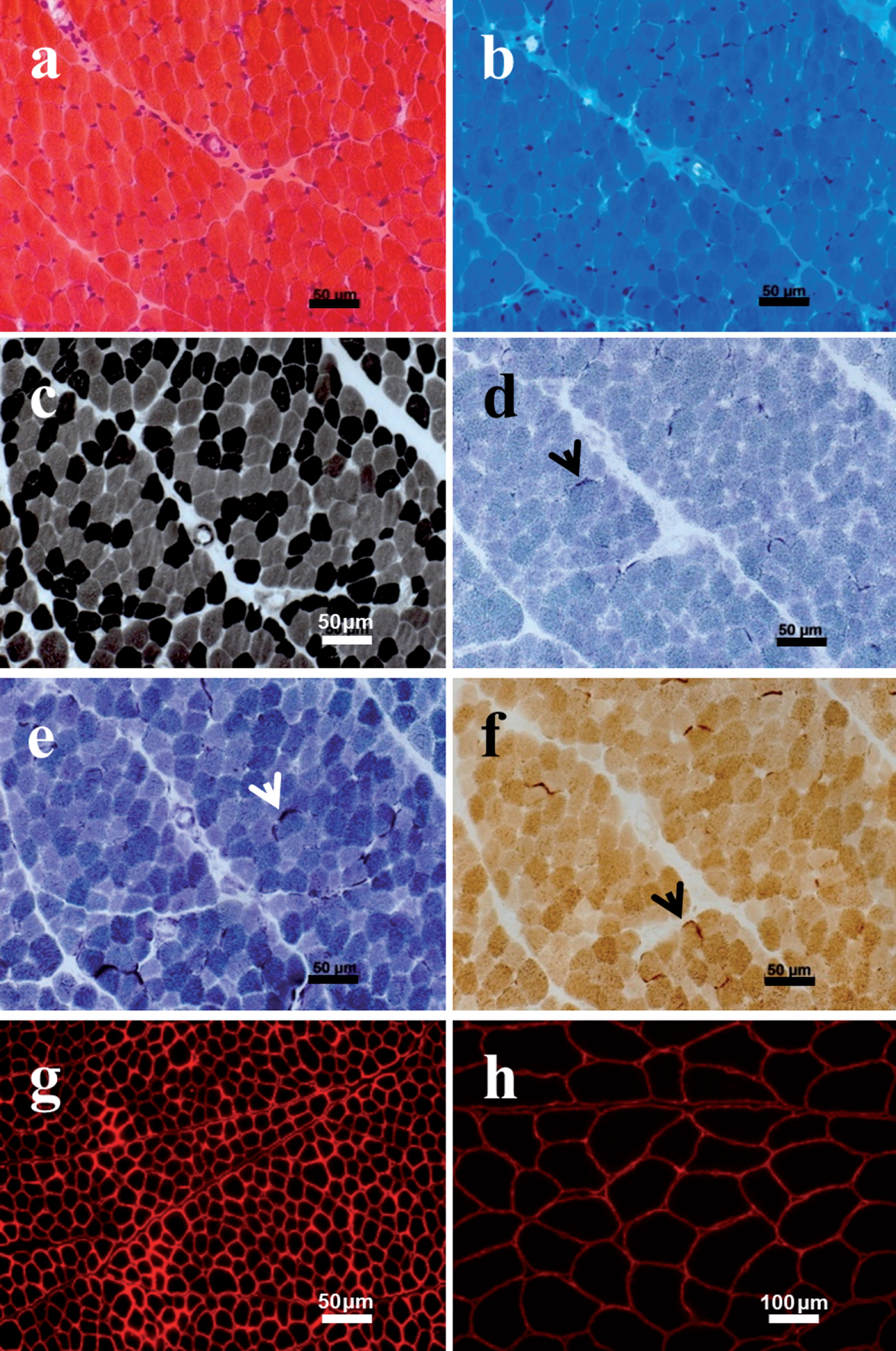
Muscle biopsy at age 2.5 years. Hematoxylin and eosin (a) and Gomori trichrome (b) stains disclosed mild fiber size variation. There were no necrotic or regenerating fibers and no endomysial fibrosis. No lymphocytic inflammation, inclusion bodies, vacuoles or ragged-red fibers were identified. ATPase staining at pH 9.4 (c) showed selective atrophy of type 2 fibers, without fiber type grouping. SDH (d) and NADH (e) staining revealed a mild increase in subsarcolemmal mitochondrial aggregates (arrows), showing positive staining for COX activity (f). Note normal immunostaining for α- dystroglycan (VIA4-1- α-DG) (g) compared with control (h).
Morbid genes encompassed in the chromosomal region duplicated in the patient.
Isoelectric focusing of serum transferrin was normal, excluding a congenital disorder of N-glycosylation (CDG). Sanger sequencing of the SIL1 gene associated with Marinesco Sjögren syndrome identified no pathogenic variants. Chromosomal analysis using array comparative genomic hybridization (aCGH) detected a large duplication affecting chromosome 11 which was then confirmed by fluorescent in situ hybridation (arr 11q13.2q14.1(66,818,265x2-66,831,965-77,781,070x3,77,815,456x2)dn). Parents were not carriers, confirming a de novo occurrence of the duplication.
Given the potential mitochondrial abnormalities initially suspected, treatment by L-carnitine and Riboflavin was started at age 3 years. Treatment was well tolerated and the patient showed improving motor function. She achieved independent ambulation at age 3.5 years, although her gait was waddling and broad-based and she was only able to walk short distances. Follow-up examinations revealed stable proximal weakness along with progressive kyphoscoliosis from age 3 years. At ages 7 and 10 she had orthopedic surgery for painful recurrent hip dislocation due to acetabular dysplasia. Adapted bracing was required from age 14 years and allowed stabilization of the spinal deformity. She had several cataract surgeries on the right eye and residual visual acuity was very low (light perception).
When last examined at age 16 years, she had stable global hypotonia and amyotrophy, proximal weakness more prominent in shoulder girdle and proximal lower limbs, and relatively preserved distal strength. Kyphoscoliosis was severe but there was no spinal rigidity (Fig. 1). The patient used a wheelchair due to important fatigue and was only able to walk short distances indoors. Intellectual disability remained important. She was able to build a few short sentences and to express relatively complex concepts but she never became fully verbal or acquired reading and writing skills. Behavior abnormalities were always present, in particular a tendency to inhibition. In the last year, she developed a depressive syndrome and auto-aggressive behavior attributed to further impairment in her visual abilities.
Periodic screening of cardiopulmonary function during follow-up revealed no abnormalities on echocardiogram or electrocardiogram, but the patient developed a progressive respiratory insufficiency. At age 5 years, forced vital capacity was moderately decreased in the upright position (68% of predicted values) with a severe drop in supine position (40%), raising the possibility of a diaphragmatic failure. By age 10, nocturnal polysomnography revealed hypoventilation with an apnea-hypopnea Index (AH index) of 30 and nocturnal hypercapnia. Nocturnal noninvasive ventilation using BiPAP was then started. At last follow-up at 16 years, forced vital capacity was 35% of predicted values (probably underestimated due to poor cooperation), and blood gas exchange was normal during the day. Gastrointestinal symptoms were also present, including episodes of severe constipation with fecalomes and signs of gastroesophageal reflux. Her skin was not markedly hyperelastic and there were not hypertrophic scars.
DISCUSSION
We report a patient with an early onset complex phenotype characterized by intellectual disability, congenital ocular and anatomical CNS involvement along with a muscle phenotype. The latter was characterized by hypotonia, proximal muscle weakness and amyotrophy, progressive and severe kyphoscoliosis, joint hyperlaxity, recurrent hip dislocation and respiratory failure. CK was normal, EMG disclosed a myopathic pattern and two muscle biopsies revealed non-specific myopathic features.
Considering the multisystem involvement and potential mitochondrial abnormalities in the muscle biopsy, a mitochondrial cytopathy was initially suspected and the patient was treated with L-carnitine and Riboflavin, which was concurrent with slow motor function improvement. Nevertheless, a mitochondrial defect was subsequently excluded by analysis of the muscle respiratory chain enzymes, southern blot, qRT-PCR of mitochondrial DNA and POLG gene sequencing. In view of the main clinical findings, MEB disease due to α-glycosylation defects was evoked. Although some necrotic and regenerating fibers were found in her first muscle biopsy, they were not observed in a second sample which showed normal α-dystroglycan immunostaining. Moreover, CK levels were normal, there was no calf or tongue hypertrophy, joint contractures or rigidity of the spine and her clinical course showed motor improvement with no progressive muscular weakness. Furthermore, a NGS-based gene panel excluded mutations in 18 known dystroglycanopathy genes.
A CGH array revealed a large duplication affecting chromosome 11 (11q13.2q14.1). The potential contribution of CNVs to a particular phenotype depends on many factors such as gene content, gain or loss of genetic material (duplication or deletion), inheritance pattern, previously recognized pathogenic CNVs in the same region, and frequency in unaffected populations. In this respect, larger deletions or duplications are more likely pathogenic since they have a higher chance of including a dosage-sensitive gene and may include a larger number of genes that cumulatively could result in an abnormal phenotype [9, 12]. Our patient’s duplication is approximately 11 Mb in length and it encompasses more than 120 genes, of which at least 41 are implicated in human disease (Table 1). These include in particular B3GNT1 associated with Walker-Warburg syndrome [13, 14], NDUFS8 or NDUFV1 associated with Leigh syndrome due to mitochondrial complex I deficiency [15, 16], the PACS1 gene causing Schuurs-Hoeijmakers syndrome [17] or SUV420H1 and DHCR7 which are reported as mutated in neurodevelopmental disorders [18, 19]. To our knowledge, no specific study on the dosage-sensitivity of these genes in cases of duplication (as opposed to haploinsufficiency) has been performed. Moreover, clinical features that argue in favor of a chromosomal disorder include dysmorphic features, intellectual disability with behavioral problems, global clinical stability and severe glaucoma. Furthermore, hypoplasia of the corpus callosum, a remarkably heterogeneous feature, has been associated with multiple rare CNVs [20, 21].
The pathogenicity of the duplication reported here is further supported by its large size, its de novo occurrence, the exclusion of mutations in candidate genes and the association of duplications in this region with a multisystem phenotype in other patients. Indeed, according to the DECIPHER v9.24 database, three patients with 7.28, 9.37 and 6.69 Mb duplications that partially overlap our patient’s duplicated region have been reported so far. These cases shared some (although not all) clinical features with our patient. All had intellectual disability. One of them (ID 250851) had structural CNS involvement, hydrocephalus, hypoplasia of the corpus callosum, plagiocephaly and prominent metopic ridge. The second one (ID280369) had cutis laxa and joint laxity, while the last one (ID 254643) had facial dysmorphism, synophrys and muscular hypotonia.
This report aims at contributing to the differential diagnosis of muscle, eye and brain congenital disease. It also highlights the importance of including systematically chromosomal studies to search for potential CNVs in the diagnostic workup of congenital muscle disease of unknown origin, in particular for complex phenotypes with multiple organ involvement including myopathic features.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGEMENT INCLUDING SOURCES OF SUPPORT
Rocío-Nur Villar-Quiles is awarded a research grant from Alfonso Martín Escudero Foundation (Beca de investigación en universidades o centros en el extranjero, Convocatoria 2017). Authors wish to thank the patient and her family for their kind cooperation. We also thank Serge Romana and Valerie Malan (Cytogenetic Unit, Paris Descartes University, Necker-Enfants-Malades Hospital) for chromosome analysis and Drs Valérie Allamand (Research Center in Myology, Sorbonne Universités, UPMC Univ. Paris 06, INSERM UMRS 974, Pitié-Salpêtrière Hospital), and Pascale Richard (Cardiogenetics and Muscle genetics Unit, Sorbonne Universités, UPMC Univ. Paris 06, INSERM, UMRS 1166, Pitié-Salpêtrière Hospital) for collagen VI studies.