
Abstract
Dogs have long been used as a biomedical model system and in particular as a preclinical proof of concept for innovative therapies before translation to humans. A recent example of the utility of this animal model is the promising myotubularin gene delivery in boys affected by X-linked centronuclear myopathy after successful systemic, long-term efficient gene therapy in Labrador retrievers. Mostly, this is due to unique features that make dogs an optimal system. The continuous emergence of spontaneous inherited disorders enables the identification of reliable complementary molecular models for human neuromuscular disorders (NMDs). Dogs’ characteristics including size, lifespan and unprecedented medical care level allow a comprehensive longitudinal description of diseases. Moreover, the highly similar pathogenic mechanisms with human patients yield to translational robustness. Finally, interindividual phenotypic heterogeneity between dogs helps identifying modifiers and anticipates precision medicine issues.
This review article summarizes the present list of molecularly characterized dog models for NMDs and provides an exhaustive list of the clinical and paraclinical assays that have been developed. This toolbox offers scientists a sensitive and reliable system to thoroughly evaluate neuromuscular function, as well as efficiency and safety of innovative therapies targeting these NMDs. This review also contextualizes the model by highlighting its unique genetic value, shaped by the long-term coevolution of humans and domesticated dogs. Because the dog is one of the most protected research animal models, there is considerable opposition to include it in preclinical projects, posing a threat to the use of this model. We thus discuss ethical issues, emphasizing that unlike many other models, the dog also benefits from its contribution to comparative biomedical research with a drastic reduction in the prevalence of morbid alleles in the breeding stock and an improvement in medical care.
Keywords
INNOVATIVE BEDSIDE TREATMENTS: CULMINATION OF LONG-TERM COOPERATIVE INITIATIVES
The international community of people that has long been involved in the fight against neuromuscular diseases (NMDs) is about to witness a major transformation in the medical management of these rare diseases 2 . Many of the innovative therapies on the market were first tested in dog models. Because of their close relationship with humans and the affective status between both, contribution of dogs is preferably referred to as cooperation. Remarkably, all the stakeholders active in research have indeed cooperated with scientists. In particular, the central role played by patient advocacy groups will be briefly reminded here.
The era of innovative treatments for rare diseases
NMDs represent a collection of ∼ 955 distinct rare entities [1], mostly inherited. For this reason, this review mainly focuses on dog models that spontaneously develop Mendelian NMDs. Although each NMD is a rare condition, the prevalence of NMDs as a group is estimated at one to three out of a 1,000 cases, which is very similar to that of Parkinson’s disease for example [2, 3]. This data supported the need for the development of orphan drugs in the last decades [4]. Biogen’s Spinraza (nusinersen) targeting spinal muscular atrophy (SMA) was the first gene (SMN2)-based antisense oligonucleotide medication for a NMD validated worldwide [5]. It has been approved by the FDA in 2016, followed in May 2019 by the FDA approval of Zolgensma, a SMN1 gene replacement therapeutic vector developed by AveXis/Novartis [6–8]. Other drugs that have been validated only in specific countries or that are still in clinical trials may follow soon [9–11]. In the last decade, 92 clinical trials targeting orphan diseases and using modern biologicals such as gene therapies or antisense oligonucleotides technology have been launched [4]. While the pharmacoeconomic threat they may pose is real, with a requested price that exceeds sometimes by tenfold the reasonable threshold of ∼200,000 € discussed by some national agencies to treat a rare disease, these modern advanced therapies open a new era in treating NMDs [12]. The cost of a gene therapy depends on several factors including the country – two-fold higher for a gene therapy targeting unresectable melanoma in U.S compared to Australia [13]-, the number of patients who will benefit the drug per year and the cost of illness to patients, families and health care systems that is calculated by adding direct medical care and indirect costs [13–15]. Using studies conducted in the U.S. and evaluating the economic impact of neuromuscular diseases [14, 15], we found that from the age of three years – average onset of the disease – to the age of 29 years-old, a patient affected by Duchenne Muscular Dystrophy (DMD) has increased direct medical costs of $805,526 and increased indirect costs – including food, travels, home and vehicle modifications and income loss – of $767,340. Altogether, additional costs per-patient over this 27-year period sum up to $1,572,866 (or 1.4 million euros). These calculated costs are likely debated during negotiations between the pharmaceutical industry and insurance systems to agree on an acceptable price for any new advanced therapy medicinal product.
Human cooperation to promote biomedical research on NMDs
The undeniable success of modern therapies represents the culmination of an experimental medical approach whose roots date into the 1850 s, with the clinical description in medical journals of neuromuscular diseases, often eponyms, published by famous founders of neurology such as Moritz Romberg, Guillaume Duchenne or Jean-Marie Charcot, among others. A century later and ten years after a first initiative in the UK, Paul Cohen, a prominent New York business leader affected by a muscular dystrophy, gathered people connected to muscular dystrophy to create a fundraising organization that became the Muscular Dystrophy Association (MDA). In nearly 70 years, the MDA has committed more than $1 billion to accelerate biomedical research and cure NMDs [16], a model that disseminated worldwide with now 74 connected associations [17]. In addition to supporting patients, many of these associations have been proactive in shaping policies and research. Indeed, they contributed to fund basic research in partnership with official research or health agencies, and promoted discovery of variants and orphan therapies for rare NMDs, often financially supporting the early preclinical or clinical steps of drug development. This is evidenced by their supporting role in dog colony programs that have been decisive in promoting feasibility of innovative gene therapies [18–20]. Vitality of rare diseases research has benefited from an improved global cooperation and collaboration among the many stakeholders active in rare diseases research, through the emergence of initiatives such as the International Rare Diseases Research Consortium (IRDiRC) founded in 2011 [21].
Cooperation of dogs among other biomedical animal models
The essential cooperation of domestic and laboratory animals have led to major medical successes in the last two centuries and led to a doubling of the lifespan of people receiving medical care [22]. Before the emergence of the rodents as popular research models back in the early twentieth century, experimental physiologists had extensively used the largest companion and farm animals. It is estimated that the discovery made by François Magendie and Charles Bell of the anterior (ventral) root of the spinal cord driving motor impulses, and the posterior (dorsal) root driving sensory impulses, required the use of 4,000 to 9,000 dogs [23]. This major discovery in neurophysiology led to the further understanding of the role of motoneuron loss in spinal muscular atrophy. Between its creation in 1901 and 1934, the Nobel prize in Physiology or Medicine awarded 13 scientists who used the dog as a model, including Ivan Pavlov in recognition of his work on the physiology of digestion, Willem Einthoven for his discovery of the mechanism of the electrocardiogram, and Frederick Banting and John Macleod for the discovery of insulin [24]. Despite such recognized evidences for conserved physiological mechanisms between humans and dogs, the putative benefit of comparative neurology was unappreciated until the end of the 1950 s. In a review dedicated to myopathies in the dog, the veterinarian Hans Meier, working at Harvard Medical School wrote: “Although there is no clear parallel in human pathology to all diseases of voluntary muscles in domesticated animals, this communication describes apparently the first instances of spontaneous myopathies in dogs microscopically identical with involvements of the striated musculature in man” [25].
Thirty years later and for the first time, the dog was presented, in a leading article in the field, as a faithful model to unravel poorly understood pathogenic mechanisms of Duchenne dystrophy in boys, reporting the lack of the same protein named dystrophin in dystrophic dogs [26]. This similarity based upon a thorough phenotypic evaluation has been highly documented and placed the Golden retriever muscular dystrophy model (GRMD) as the gold standard in systemic, integrative evaluation of modern therapies, complementarily to the mdx mouse model that is preferred in early preclinical proof-of-concept studies [27, 28]. Faithful complementarity of the continuum of models relies on both their internal and external validity. Internal validity represents the scientific robustness of a study’s design, conduct analysis and reporting, while external validity is the extent to which research findings in models can be reliably applied to humans [29].
Although the validity of animal models is a constant matter of debate and criticisms [30, 31], the use in the NMD field of complementary models such as the mouse and the dog has already proved its efficiency for the treatment of two of the most devastating diseases in children, namely the X-linked recessive Duchenne dystrophy and centronuclear myopathy. In both cases, intravascular infusion of antisense oligonucleotides (ASO) or AAV-mediated gene/ASO constructs allowing either the reframing of DMD or the expression of a functional cDNA encoding microdystrophin or MTM1 had been conceived and tested in mice [32–34], then validated in dogs that offered a proof-of-confidence in a larger animal model [18, 35–40], then paving the way for clinical trials in patients [9, 41]. Last year, therapeutic edition of the DMD gene was successfully achieved in a DMD dog model, after systemic delivery of the AAV-vectorized CRISPR/Cas9 machinery [19], providing strong evidence that genome edition is an actionable, efficient, presumably safe and unlimited therapeutic strategy.
THE UPDATED LIST OF DOG MODELS OF NMDS
Among the numerous hereditary neuromuscular diseases characterized in dogs, many represent relevant models for human NMDs (Table 1). As of July 2019, the list included 45 models with mutations in 25 nuclear disease-causing or modifying genes, of which 21 were identified as NMD-causing genes in humans (Table S1). Additional models of acquired NMDs are provided in Table S2. [42–88]
List of dog models for inherited NMDs. The groups are those used in the classification of human NMDs ([91] and www.musclegenetable.fr). Genome annotation may have been updated since the initial report; in this case, the present Ensembl annotation was used as a reference (CanFam3.1) explaining, for example, that number of the exon carrying the mutation may differ from the original report. No dog model has yet been reported in groups 4 and 6. An asterisk after the “Year of mutation identification” indicates that the first clinical description of the disease anticipated the genetic characterization. In this case, the Year of first clinical description and the associated reference are provided in an extended version of the Table, available online as Table S1. Groups 10 (hereditary cardiomyopathies), 13 (hereditary ataxias) and 15 (hereditary paraplegia) were voluntarily omitted
All dog models display clinical signs, severity and time-course of the disease that are highly similar to human conditions and in many cases, more similar than mouse models. Some examples include Landseer dogs with variants in the COL6A1 gene, models for the Bethlem myopathy or Ullrich congenital muscular dystrophy [56], DMD dogs with variants in the dystrophin (DMD) gene, models for Duchenne muscular dystrophy [48] and dogs with variants in the NDRG1, ARGHEF10 or GJA9 genes, models for Charcot-Marie-Tooth (CMT)-like neuropathies [80–83]. In all these examples, mouse deficient models express a milder clinical phenotype, as for example described in mdx or Col6a1-KO mice [89, 90].
The dog is also the most studied mammalian model that spontaneously develops human-like NMDs. For example, it was shown that a variant in the superoxide dismutase 1 (SOD1) gene segregates in the Boxer, Pembroke Welsh Corgi and German shepherd breeds, producing a missense E40K mutation in the protein. In homozygous owners’ dogs, E40K homodimers aggregate in motoneurons that die, eventually leading to an ALS-like late onset degenerative disease that fully parallels the time course of human amyotrophic lateral sclerosis (ALS) [77].
The 25 genes identified in dogs represent less than 10% of the 308 human genes harboring known NMD-causing variants, after excluding groups 10, 13 and 15 of the monogenic NMDs classification [91] which are outside the scope of this review. Characterization of further spontaneous dog models is ongoing by several teams, including ours, and will lead to an expanded list of variants and genes.
THE DOMESTICATED DOG, A GENOMIC MIRROR OF MAN’S RECENT EVOLUTION
To address complex questions such as the respective function of NMD-related paralogous genes that resulted from evolutionary duplications, we still need to investigate mechanisms in complex vertebrate organisms. Furthermore, the requirement of integrative mammalian models as close as possible to humans is mandatory in order to evaluate how these duplicated genes may be used as therapeutic targets, for example in human patients affected by spinal muscular atrophy [5], or to precisely assess pharmacokinetics of a therapeutic molecule in muscles. Researchers need models with vascularized and innervated muscles, in a body metabolizing drugs in a liver, eliminating metabolites either in urine formed and modified in kidneys or in feces after bile salts conjugation; a body breathing, with circulating blood maintaining homeostasis, interacting with a constantly changing internal or external environment, and able to respond accordingly with fully functional immune, endocrine and nervous systems.
Evolutionary species divergence and functional convergence
The carnivore order belongs to Laurasiatheria, the fourth clade of eutherians, while rodents, rabbits and primates, including humans, belong to the third clade of Euarchontoglires (Fig. 1A). After studying the chronological divergence of orders through speciation, one may deduce that the younger common ancestry between mice and humans makes the mouse a closer, more reliable biomedical model. Strikingly, alignment of unique sequences in the euchromatic portions of the dog, human and mouse genomes revealed on the contrary that dog shares more orthologous ancestral sequences with human, exceeding by 500 Mb the length of ancestral orthologous sequences shared between human and mouse [92]. To investigate whether this higher dog-human relatedness would also be relevant to genes specifically related to the neuromuscular system and to include other putative models, we selected 22 genes with known NMD-causing variants, and compared the exonic, 5’ and 3’ untranslated regulatory regions from the mouse, rat (Euarchontoglires), dog and pig (Laurasiatheria) reference genomes with the human sequences. The percentage of similarity with human sequences was compared between two species and revealed that the dog and pig nucleotide sequences are significantly more similar to that of human, than the mouse and rat (Fig. 1B). While this result may only represent the tip of the iceberg, it highlights that for a given function, proximity in the phylogenetic tree is not necessarily associated with a higher similarity in genome sequence and thus, in functional pathways [93]. In other words, distant species such as the dog and human are eventually genetically closer, presumably because mechanisms such as functional convergence and shared environmental pressure shaped genomes in the same way [94, 95].
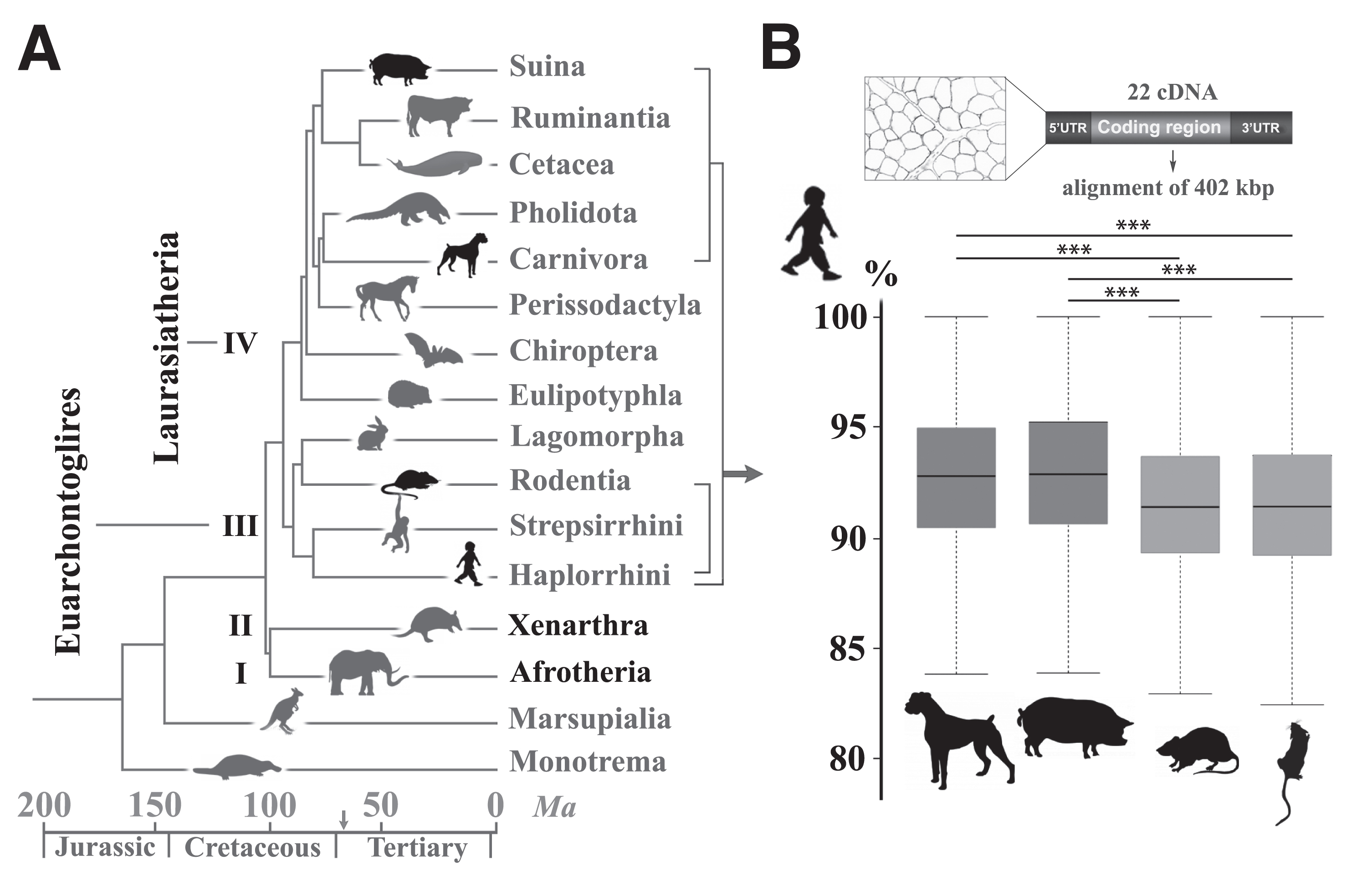
Nucleotidic sequence similarities in neuromuscular disorder-causing genes between humans and three eutherian models. (A) Timetree of mammalian diversification showing the four clades of eutherians (I to IV, black names). Rodents (Rodentia order) and Humans (Primates) belong to the Euarchontoglires clade while dogs (Carnivora) and pigs (Suina) belong to the Laurasiatheria clade. Other orders are reported in grey. Note the extreme diversification that occurred 66 million years ago (Ma) during the Cretaceous–Palaeogene fifth mass extinction event, represented on the time scale by an arrow. (Adapted from [259]). (
Domestication of the dog
These two mechanisms have been triggered by the domestication of animals, and dogs were the first ones. Dog domestication is thought to have occurred around 35,000 years ago, at the junction between the end of the Middle Paleolithic Age – the era of Neanderthal humans, – and the Upper Paleolithic Age during which only the modern Homo sapiens existed. Molecular analyses have clearly established that modern dogs all derived from a common, now-extinct ancestral population of wolves. The geographical origin of this domestication is a matter of active research, but presumably happened simultaneously in distinct locations such as Europe, the High Arctic and East Asia [96]. A second stage of domestication, around 11,500 years ago, was concomitant with the switch towards cultivating wild cereals and legumes during the Pre-Pottery Neolithic Age. Cooperation between dogs and humans increased, and in addition promoted domestication of other animal species that became our modern farm animals, starting 10,500 years ago [97].
Genomic signatures of domestication and coevolution
In that time, the phenotype of dogs diverged from ancestral wolves, including morphological, behavioral and functional adaptations underpinned by favorable, selected genomic variations that became molecular signatures of domestication, grouped within 30 regions corresponding to 1% of the genome and encompassing 100–300 genes [94, 95]. These regions are enriched in genes playing roles in brain function, gamete recognition, ossification, neuromuscular junction formation, starch digestion and metabolic processes. The most illustrative and documented example is the copy-number increase in the amylase (AMY2B) gene in response to the high-starch diet progressively introduced by farmers, which began 7,000 years ago and anticipated the development of the agriculture-based civilization expansion in Europe [98]. Of note, comparative analysis of orthologous gene pairs between dog domestication signatures and human genome regions identified in scans for positive selection, led to the identification of 32 genes with paralleled recent evolution. For example, this was highlighted by a similar enrichment in the two ATP-binding cassette transporters superfamily ABCG5 and ABCG8, both involved in the selective transport of cholesterol [95].
Globally, this long-term coevolutionary process has shaped genomes of modern dogs and humans in a way that favors highly-similar physiological and pathological mechanisms between the two species. Thus, the dog represents a unique genomic mirror of human molecular evolution with a genomic and environmental complexity that overcomes a model induced by a single genome mutation at a locus of interest. This is likely the reason why dogs affected by diseases, and in particular NMDs, mimic human molecular pathogenic mechanisms [60], and often better than the undoubtedly useful and complementary mouse models [99].
The overall history of the dog, from its domestication to its modern relevance in comparative medical genetics and translational medicine, is presented in Fig. 2.
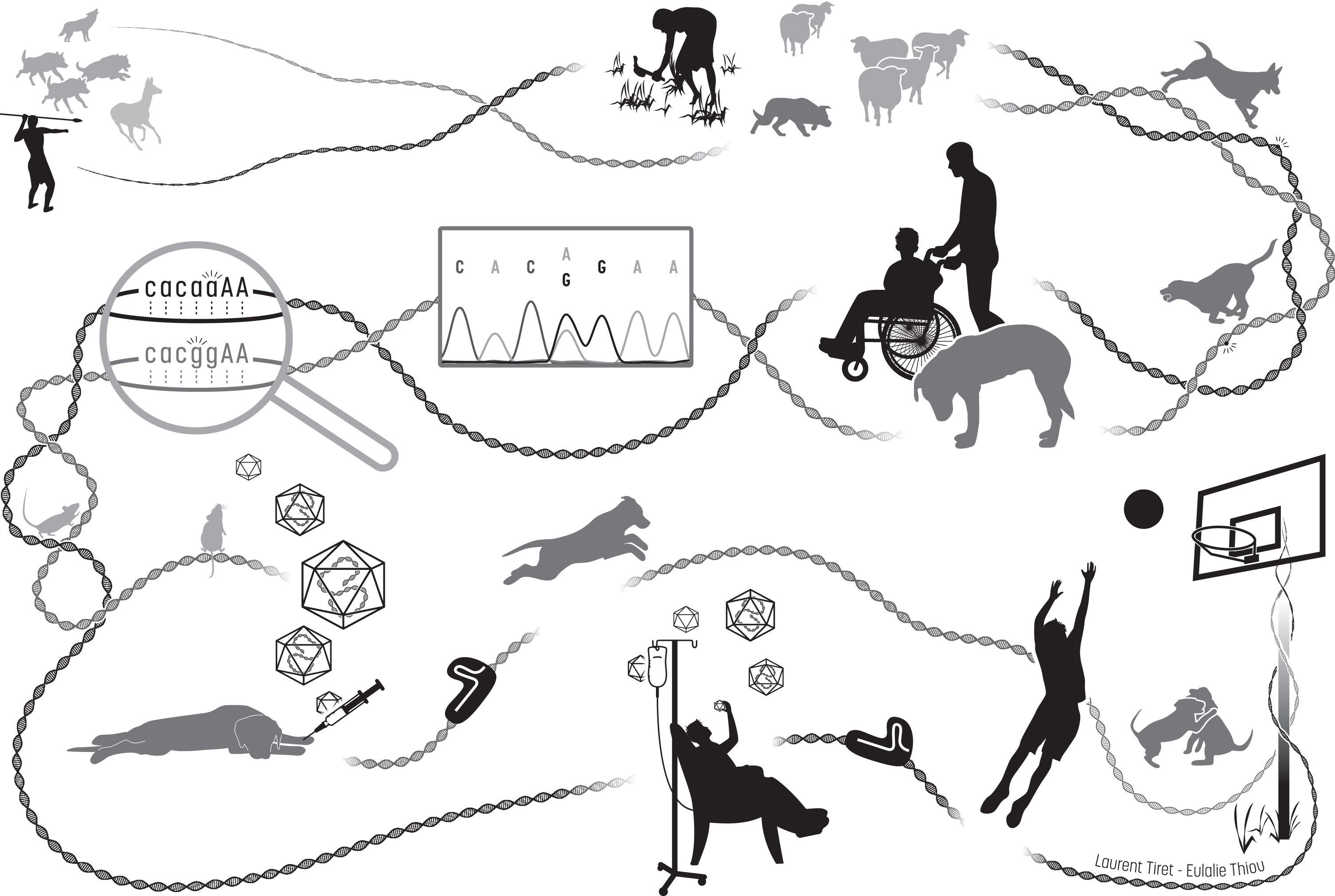
Illustrated history of the intermingled Humankind and dog recent evolution, from domestication to comparative medicine. From the upper left corner, then along the DNA path: in prehistoric paleolithic times, Homo sapiens ancestors and grey wolves which are dogs’ ancestors may have developed similar social abilities for cooperative problem solving, synergizing in convergent actions such as hunting. Between around 35,000 years ago and the beginning of the Pre-Neolithic starting roughly 11,500 years ago, humans and domesticated dogs achieved more and more cooperative tasks such as protecting herds of other domesticated farm animals. Sharing their daily life and environment resulted in common genomic signatures. By exerting new forms of selection pressure on the dog’s genome, human evolution resulted in many convergent physiological mechanisms. Over the last 300 years, phenotypic diversity increased in dogs following a sustained accentuated artificial selection of desirable traits spontaneously emerging in domesticated dogs, leading to the creation of breeds that are genetic isolates. This unfortunately led to the rapid spread of unwanted breed-specific disease-causing variants that also spontaneously happened, and in particular favored homozygosity of loss-of-function recessive alleles resulting in the emergence of hereditary disorders, including those affecting the neuromuscular system. Dysfunction of convergent physiological mechanisms lead to highly similar pathogenic mechanisms in patients and affected dogs, which are thus relevant spontaneous clinical and molecular models. In the last two decades, comparative medical genetics has allowed to identify 290 human-like disease-causing variants in 190 genes, as illustrated here with the autosomal recessive mutations identified in the same intronic acceptor site of BIN1 in human patients (ag=> aa) and affected Great Danes (ag=> gg) that display a highly similar, rapidly progressive congenital myopathy [60]. Comprehensive, longitudinal characterization of dog diseases helps establish a chronological list of quantified parameters, further used as outcome measures to evaluate in preclinical trials the relevance of innovative therapeutic strategies, such as virally-vectorized delivery of genes, oligonucleotides or the CRISPR/Cas9 machinery driving genome edition, previously shown to be effective in mice. Once validated in the large mammalian dog model, the proposed treatment can be assessed in patients enrolled in clinical trials. Robustness of this biomedical continuum in the myology field has recently been exemplified by the AAV-mediated MTM1 gene therapy [38].
Parallel evolution between the dog and human immune systems
When developing cell or gene therapy products for NMDs, an important bottleneck is the immune response that can be directed against the vehicle (e.g. donor cell, AAV vector), the transgene, or other components of the therapeutic approach such as the Cas9 protein in genome edition strategies. The mouse model greatly helped unraveling several common features of the adaptive immune system in mammals, like the regulation of antibody synthesis [100]. However, it represents a weak model for mechanisms that are more specifically dependent on divergent features [101]. For example, maintenance of memory T cells have evolved in response to coevolution of life expectancy [93], and in this regard the canine immune system more closely resembles that of the human, including in its developmental steps. Notably, unlike rodents and similarly to humans, the immune system of the dog is competent before birth and pursues its maturation postnatally [102]. Such similarities keep making dogs suitable and attractive models in many immune-related fields such as in bone marrow transplantation and graft-versus-host disease management, oncology, immunologic disorders or gene therapy of hemophilia [103–106]. Relative to human acquired NMDs, it was observed that infiltrates in canine myositis are of similar nature to those observed in human patients [107]. In myasthenia gravis, the most frequently diagnosed canine NMD, production of auto-antibodies in affected dogs mimics that of human patients [108, 109]. Finally, in several acquired canine NMDs, several MHC-related risk factors that are similar to humans have also been identified [110–112]. Thus, it is likely that dogs are good models to study the human-like immune response involved in the global adaptive strategy following onset of an NMD or a therapy. In the emerging field of AAV-driven gene therapy, studies performed in GRMD dogs have evidenced strong inflammatory response following vector injection either linked to the vector or to the transgene and promoter, revealing a decreased immunological tolerance compared to mice, hence providing opportunities to better anticipate immune reactions in humans [113–115]. Presumably as a consequence of biological convergence in evolving systems, dogs seem to share innate immune system signatures with humans. This was exemplified by a study showing that in the two coevolving human and dog species, and contrarily to mice or macaques, the AAV6 capsids interact with galectin 3 binding protein in serum, form aggregates and lower efficiency of this serotype to reach muscle after systemic delivery [116].
THE OWNER’S DOG, A SPONTANEOUS GENETIC MODEL FOR COMPARATIVE CLINICAL RESEARCH
In industrialized societies, the modern dog is often a family member and, for this reason, has access to quality medical care, diagnosis and treatment [117]. This new view of man’s best friend has partly accounted for the recent fast evolution of veterinary medicine. Veterinary specialties have emerged, organized into colleges and societies, and rapidly progressed to reach a high level of expertise [118, 119]. That is also the case for veterinary neurology, which has implemented techniques of neurological examination, exploration and pathology, adapted from human neurology, in a comparative diagnosis approach. This holds particularly true for NMDs of companion animals.
Comprehensive identification of spontaneous cases in neurology clinics
Clinical examination
The canine diagnostic approach is very similar to the one used in human patients with NMDs [120, 121]. First, the patients’ history (age at onset, sex, family history if available, breed) is taken, and a comprehensive neurological examination is performed, including general observation of the dog, detection of gait and postural abnormalities, assessment of postural reactions, reflexes, muscle tone, and sensitivity/nociception evaluation [122, 123].
Paraclinical examination
Medical biology. The serum CK measurement is part of the first paraclinical investigations and is routinely accessible to veterinarians. Other paraclinical investigations include routine biochemistry, metabolic, endocrine and autoantibodies screening [124].
Muscle, nerve and neuromuscular junction electric properties. The implementation and wide usage of electromyography (EMG) in veterinary neurology has allowed for a step forward into the diagnosis of NMDs in our companion dogs. EMG is performed on anaesthetized dogs, and first evaluates the presence of abnormal spontaneous muscle activity. Sensory and motor nerve conduction studies are also performed after nerve electrical stimulation. Proximal nerve and nerve roots can be tested through F-waves and cord dorsum potential recordings. Finally, the neuromuscular junction can be assessed by analyzing compound muscle action potentials after repetitive stimulations, a decrement being evocative of a myasthenia, in complement with a positive response to neostigmine challenge. As in human patients, EMG is thus an essential diagnostic tool to approach the diagnosis of NMDs [125].
Muscle and nerve microstructure. Another important diagnostic tool corresponds to the muscle or the nerve biopsy that are easily performed in dogs: given their large size, a significant amount of tissue can be sampled without any deleterious consequence. In this field again, specialized veterinary pathologists have developed a high degree of expertise and created reference centers, allowing for an accurate diagnosis supported by comparative pathology [124, 127]. The deployment of such a neuromuscular evaluation toolset has allowed for the clinical diagnosis of many canine neuromuscular conditions similar to human NMDs, ranging from acquired diseases including inflammatory myopathies, myasthenia gravis and Guillain-Barré syndrome, to hereditary myopathies, myasthenia, neuropathies and motor neuron diseases. Most of the mutations found in dogs with phenotypes mimicking the human condition have been found in human disease-causing genes (Tables 1 and S1).
Genomic and genetic tools for the identification of disease-causing variants
The use of models in preclinical trials assessing modern biologicals relies on their fine and essential characterization at the molecular level. The first dog NMD-causing variants were identified in the early 90 s by comparative analyses focusing on the disease-causing genes in humans [42, 69]. Significant advances resulted from the availability of mapping genomic tools such as the combination of polymorphic microsatellite panels and linkage analysis software led to the identification of gene variants that had not been previously linked to NMDs (for example in DNM1 [86] or HACD1 [61, 128], the latter being further identified as a causative gene in human NMD [129, 130]). The toolbox was dramatically improved by the sequencing and annotation of the canine genome [92, 131], initiating the development of high density single nucleotide polymorphism (SNP) microarrays and in particular the 170,000 SNP (170 k; [132]) that has been mostly used in genome-wide linkage or association studies in canine NMDs (for example in [75, 85]; Table S1). The availability of a steadily improvement of the dog genome annotation [133] also prompted association analyses using massive parallel sequencing of the genome, exome and neuromuscular tissue transcriptomes of affected dogs. It allowed researchers to identify causative variants with a constantly diminishing number of enrolled dogs (Table S1). In this regard, the most illustrative example is the identification of the DMD modifier variant in the JAGGED1 gene, elegantly pinpointed with only two escaper dogs and using an analysis pipeline including a genome-wide association study followed by sequential linkage, RNAseq and whole genome sequencing analyses [43].
THE RESEARCH DOG, A RELEVANT TRANSLATIONAL MODEL
Eight NMD-causing variants were amplified and maintained through the development of experimental colonies worldwide (Table S1). They are mainly being used to characterize pathogenic mechanisms or evaluate innovative strategies in preclinical trials, and they are models for GSD IIIa [134–136], DMD [19, 39] and centronuclear myopathies [38, 138].
Comprehensive functional evaluation and disease natural history
Long-term longitudinal follow-up studies
The long-term follow-up of dog models helps answer crucial questions for human applications.
First, once identified, the disease-causing variant can be used to genotype pups even before they start expressing clinical signs. By monitoring pups born from experimental litters, it is therefore possible, for example, to identify the earliest histological signs of the disease, which may be different from the major modifications observed at the time a biopsy is performed in a patient, months or years after the onset of the disease. Indeed, due to a large number of internalized/centralized myonuclei, dogs with mutation in HACD1 gene have been initially classified in the group of centronuclear myopathies [61, 128]. Yet, presymptomatic evaluation of muscles from genotyped healthy-looking pups revealed hypotrophy of some myofibers, which prompted focused comparative analyses that eventually led to the identification of a myoblast fusion defect [137]. This provided new clues for innovative treatments, knowing they are mostly being developed from newly deciphered pathogenic mechanisms [4].
Another remarkable example is the long-term effect assessment of gene therapy strategies. In XLMTM dogs, a 4-year follow-up after gene therapy has shown persistence of the functional improvement following AAV-MTM1 therapy, though the amount of AAV genomes progressively decreased over time in muscles [139]. In a similar way, a long-term follow-up (more than 4 years) of GRMD dogs treated with an AAV-driven exon skipping strategy showed that the AAV vectors were rapidly washed out from treated muscles, resulting in a progressive decline of the quasi-dystrophin expression, probably due to the persistence of muscle degeneration driven by the few remaining dystrophin negative fibers [35]. These essential results for an efficient translation of AAV-based gene therapy strategies underlie the necessity to propose complementary approaches or new strategies to help maintain the initial level of transgene expression.
Not only can the dog permit long-term evaluations, but it also allows scientists to longitudinally and iteratively follow the disease course during months or years, and further evaluate accurately the long-term outcome of a treatment. The dog has a level of interaction with humans which allows developing several non-invasive functional assessment methods often without requiring any sedation. This allows for iterative testing, because dogs cooperate with the experimenter and are able to quickly learn simple tasks.
Functional evaluation methods in NMD models
Gait tests.
Use of the 6-minute walk test and timed motor performances: Locomotion impairment is a constant hallmark of NMDs described in dogs, which is expected to improve if a systemic treatment is efficient. Therefore, efforts have been undertaken by teams using dogs for preclinical trials to establish gait evaluation tools which could be used to quantify and monitor gait impairment over time. In order to parallel outcome measures used in clinical trials for human NMDs, the widely used 6-minute walk test (6MWT) was evaluated in two dog models, the GRMD and the HACD1-CNM dogs [140, 141]. The principle of the test is roughly the same as in humans (i.e. an evaluation of the maximal distance that can be walked by the dogs over 6 minutes). It however differs de facto from the original 6MWT, since the test in humans is based on standardized instructions given to the patient before the test, which cannot apply to dogs, and besides, dogs are not restrained to the walking gait. In both models, the diseased dogs walked a shorter distance than healthy dogs and the test was evaluated as feasible with minimal training requirement (leashed walk). Other advantages include the minimal technology required to perform this test, allowing for an easy set-up by any team working on dog models. Some pitfalls of this test include the lack of discriminating power, the lack of clear correlation with other functional tests in GRMD dogs [140], and the fact that this test does not specifically address gait, but rather a mix between locomotor, respiratory and cardiac performances. A timed running test was alternatively used in DMD dog models, consisting of a 15-meter timed walk or run, and this test was able to demonstrate an improvement of the performance after gene therapy [18].
Use of gait analysis gold standards (kinematics, kinetics): Dog models of NMDs have also been evaluated using gold standard methods of gait assessment, i.e. kinematics and kinetics-ground reaction forces measurement [142–145]. Kinematics, based on a high-frequency video acquisition of dogs wearing joint reflective markers during gait, highlighted reduced hock joint range of motion, less flexed hock, and more extended stifle in GRMD dogs [142]. These results were confirmed by another study, while performed without placing markers on joints, which also showed decreased ranges of motion of other joints, and found the carpus to have a smaller flexion angle and a larger extension angle in GRMD dogs [143]. Conversely, in the XLMTM dog model, hock and stifle kinematics were not modified [145], demonstrating that this method cannot be used as a universal tool to evaluate gait in dogs with any NMD. Ground reaction forces were measured only in one study involving GRMD dogs, showing an elevated vertical force increasing with age, as well as a decrease of the cranio-caudal force, unfortunately without any direct comparison to healthy dogs [144]. The equipment required for ground reaction force measurement (force platforms) is very specialized and expensive, and most likely the reason there have been few studies investigating this gait analysis method in dogs with NMDs.
Alternative gait analysis methods (instrumented carpet, 3D-accelerometry): Alternative gait evaluation methods have been proposed, to circumvent the high cost and equipment level required for gold standard gait analysis. An instrumented carpet consisting of a 7-meter long pressure sensitive walkway, coupled to software which calculates spatiotemporal gait indices, was successfully used in the XLMTM dog model. This test is reported to be easily feasible with XLMTM dogs and was able to quantify gait disabilities in animals that exhibited decreased velocity, stride length and increased stance time [146]. Probably not restricted to the XLMTM, these markers of gait disability were sensitive to treatment, since they improved significantly following systemic gene therapy [38]. In dystrophin-deficient dogs, gait evaluation using three axial-accelerometry has been proposed and used by several groups [147–149]. In the most recent study, three-axial accelerometers were positioned on the back at thoracic and lumbar levels, providing regionalized acceleration magnitude and angular velocity data, which for some of them nicely correlated with other markers of disease severity [149]. Alternatively, a unique three-axial device positioned near to the center of gravity at rest has been proposed, taking advantage of the fact that tri-axial accelerations at the center of gravity reflect exerted forces during gait. This method has been used in GRMD dogs, in which it was able to highlight and quantify many gait abnormalities, including decreased velocity, stride length and frequency, total power of accelerations, and increased relative medio-lateral power [147]. These gait indices, most altered very early on in the disease course, were found to correlate with other indicators of disease severity. This test was simple to perform, well tolerated and thus could be used to iteratively evaluate the animals, allowing for detailed longitudinal studies [150]. Given the numerous gait indices found to be modified in GRMD dogs, a statistical method intending to combine all these indices and more clearly conclude on the effect of a treatment was proposed [148]. Importantly, 3D-accelerometry based gait analysis was revealed to be sensitive both to a moderate treatment effect [148, 151], and to a strong treatment effect [39].
Respiratory function tests. Respiratory muscle function can also be assessed in dog models of NMDs. In both XLMTM and GRMD dogs, Tidal-breathing spirometry, performed using a pneumotachometer linked to a facemask, or respiratory inductance plethysmography (RIP) allowed precise quantification of respiratory muscle disability. Interestingly, the observed abnormalities were different between both diseases. In XLMTM dogs, a thoraco-abdominal asynchrony and a decreased peak inspiratory flow (PIF) with low response to doxapram were measured, signaling weakness of the diaphragm. Again, these markers of diaphragmatic weakness were greatly improved after gene therapy [38, 152]. In this model the respiratory test was performed under general anesthesia and with a doxapram challenge. On the contrary, the respiratory tests were performed on awake GRMD dogs and were well tolerated. They revealed an increased peak expiratory flow (PEF), an increased PEF/PIF ratio, and increased ratios of PEF on late expiratory flows [153]. All these abnormalities are due to an increased diaphragm stiffness and compensatory recruitment of muscles, notably from the abdominal wall [154]. Whether these respiratory mechanics abnormalities measured either by Tidal-breathing spirometry or RIP would be sensitive to the effect of a treatment in the GRMD dog model is still to be investigated.
Clinical scoring. Inspired by scales used in patients with NMDs, some scoring grids have been proposed for dog models, based on the clinical signs observed and on their severity. In DMD dog models, the evaluated items focused on motor aspects such as the gait, posture, contractures, or on digestive and respiratory aspects such as drooling, dysphagia or dyspnea signs [155, 156]. These clinical scoring grids were used and found able to detect the effect of a treatment, together with other functional improvement clues, following gene or cell therapy [18, 157]. In the same way, a neurological assessment score was proposed for the XLMTM dog model, including gait evaluation, drop of the jaw due to muscle weakness, muscle atrophy and respiratory distress [158]. Again, this scoring method was efficient in detecting the effect of a treatment, since the score was improved in dogs systemically treated by gene therapy, in a dose dependent manner [38]. A disease stage grading has also been proposed for dogs affected by a SOD1-related degenerative myelopathy [159, 160].
Cardiac function evaluation. Many NMDs do not only affect skeletal muscles, but also the myocardium. Among these, DMD leads to a late onset cardiomyopathy, characterized by an occult phase during which myocardial fibrosis appears and progresses, ultimately leading to a dilated cardiomyopathy. Given great advances made in the respiratory management of these patients, cardiomyopathy has become a more frequent cause of death and is therefore a major therapeutic target. Dog models of DMD also mimic this aspect of the disease. Initial pathological observations, observed between 6 and 12 months of age, include cardiomyocyte degeneration, mineralization foci and fibrosis of the left ventricular free wall, which progresses after 12 months to become prominent, spreading to the whole myocardium, and fatty infiltration [161]. Due to the pathological similarities between dystrophin-deficient dogs and DMD patients, including in the timeline of the cardiac involvement relative to locomotor and respiratory impairments, many efforts have been undertaken to describe and quantify the canine DMD cardiomyopathy, using the same tools as in DMD patients.
Electrocardiographic studies: First, several electrocardiographic (ECG) studies have been performed in GRMD or other colonies of dystrophin-deficient dogs. Most of these studies used classical ECG recordings in non-sedated dogs, and highlighted elevated heart rate, deep Q-waves, shortened PR interval, and sporadic premature ventricular beats [162–164]. The deep Q-waves were shown to be improved after gene therapy, indicating that they could serve as an indicator of treatment efficacy [165]. More recently, long-term Holter ECG was used in DMD dog models, showing more diverse arrhythmias, still with premature ventricular beats, but also with premature atrial contractions and non-conducted P-waves [54, 166]. Interestingly, marked long-term arrhythmic profiles were observed in GRMD carrier female dogs, thus strongly suggesting, together with the observation of significant pathological lesions in the myocardium, that carrier bitches faithfully mimic the cardiac disease described in some human carrier females [167, 168].
Echocardiographic studies: Like in DMD patients, conventional echocardiography performed in non-sedated dogs allows the detection of decreased ejection fraction, fractional shortening, left ventricular free wall and septum thicknesses, and an increase of the end-diastolic and end-systolic left ventricular internal diameters in DMD dog models aged more than one or two years [162, 169–171], which is consistent with the evolution of a dilated cardiomyopathy. While late onset, like in DMD patients, these features have been shown to be treatment-sensitive since they improved after pharmacological therapies [170, 171]. Earlier in the disease course, conventional echocardiography allows only the identification of hyperechoic lesions linked to mineralization and fibrosis [162, 163]. In order to increase the sensitivity of echocardiography at earlier timepoints, more advanced methods have been evaluated, such as speckle tracking imaging, which allows the measurement of regionalized myocardial deformation. In the occult phase of dilated cardiomyopathy, DMD dog models were found to have a decreased early-diastole peak radial strain rate in the posterior segment [172]. Also, in an attempt to identify early echocardiographic markers looking at myocardial deformation, Doppler tissue imaging (DTI) has been proposed. This method revealed markedly decreased endo-epicardial gradient of velocities in young GRMD dogs (six months of age) with normal conventional echocardiography, allowing for an early detection and monitoring of the cardiomyopathy, in young animals as well [173]. This marker was shown to be treatment sensitive since it normalized together with conventional echocardiographic indices following a pharmacological treatment [170].
Cardiac MRI: During the last years cardiac MRI has become part of the standards of care for DMD cardiomyopathy, and is of particular utility, not only to monitor cardiac function in DMD patients, but also in the early detection of myocardial fibrosis, which precedes the decrease of the ejection fraction [174–176]. Given this growing interest in cardiac MRI, GRMD dogs were also evaluated using this technique, requiring general anesthesia in canines. Early ventricular segmental dyssynchrony was observed in GRMD dogs, and tagged sequences revealed decreased peak circumferential strain, which were improved following gene or pharmacological therapies [169, 177]. Finally, cardiac perfusion MRI has been assessed using a gadolinium injection, revealing increased perfusion but no myocardial scar in a small group of DMD dog models [178]. However, another study reported on the presence of both fibrosis and fatty infiltration in the myocardium using Dixon sequences, and hyper-enhanced lesions using late gadolinium enhancement [48].
Skeletal muscle imaging.
Magnetic resonance imaging and spectroscopy of skeletal muscles: In human NMDs, magnetic resonance imaging (MRI) is a widely used method of muscle imaging, allowing for comprehensive characterization and mapping, and non-invasive follow-up of muscle shape over time. In dog models of NMDs, even if iterative muscle biopsies can be more easily taken than in human patients, it only gives information on a small piece of one muscle among hundreds. When assessing treatment outcomes, MRI represents an invaluable tool to conclude in a more reliable manner on the shape of several muscles, and follow them over time, during or without treatment. Indeed, MRI has also become a key tool in the evaluation of dog models of DMD. Both hindlimbs (thigh and leg) and forelimbs (forearms) have been studied, and a common observation between all the studies performed in dystrophin-deficient dogs is the T2 signal hyperintensity and heterogeneity, reflecting patchy distribution of necrotic and inflammatory areas. This has been quantified on images by either increased T2w/T1w, T2wFS/T1wFS, T2w, T2 relaxation time, CHESS-T2w or decreased PDw/T2w [155, 180]. The patchy distribution of such T2 hyperintensities has been quantified using indices derived from the standard deviation of the T2w signal [155, 180]. Another common finding on muscle MRI images from dog models of DMD is the markedly increased post-Gadolinium enhancement, reflecting both necrosis and increased extracellular volume. These indices have been used to monitor treatment effects and have proved to be useful in detecting improvements in muscle shape. After a local or locoregional treatment, the treated limb was compared to the contralateral untreated one, and a reduction of the T2 hyperintensity, heterogeneity and of the relative enhancement following different approaches of gene therapy has been demonstrated [35, 39]. In the same way, the T2 hypersignal was decreased after systemic gene therapy [18, 181].
New advances in MRI image acquisition and processing allow for the measurement of T2 maps, water- and fat-maps (all increased in GRMD dogs), and texture analysis allowing for a 3D analysis of the distribution of lesions, quantified by indices such as SLI (small lesions index), HI (heterogeneity index) or entropy [182–184]. Again, these new indices were sensitive to the effect of a treatment, since they were shown to improve after pharmacological therapy [185]. All these indices have also proven being useful to monitor the deleterious effect of a treatment, such as inflammatory response after AAV injection, evidenced by an increased T2 [113, 186]. Other indices useable in DMD dog models include an increased T1 value and T1w heterogeneity [180]. On older dystrophic dogs, MRI reveals fat infiltration, but at later stages than those usually assessed in preclinical studies [156, 179].
A basic question about MRI is the correlation between image-derived measured indices with histological lesions. Dogs with NMDs represent a key to answer this important question. Some studies have investigated this question and tend to show a correlation between the amount of necrosis and inflammation in dog muscles and the T2 increase [113, 186]. Although a recent study proposed a method to accurately address this challenging question [187], extensive studies on this topic are still lacking. In the same way, dog models may serve to characterize correlations between MRI and muscle function. Recently, a positive correlation has been established between T2 hyperintensity of some muscle groups and gait impairment assessed using accelerometry [149].
MRI also allows for muscle volume measurement, demonstrating a decreased muscle volume in dog models of DMD, with the exception of specific muscles such as the Sartorius cranialis, which undergoes hypertrophy in GRMD dogs [188]. MRI-based calculation technique has been used to monitor an increased muscle volume after either therapeutic intervention or crossbreeding to inhibit myostatin [181, 189].
Finally, the imaging of the GRMD diaphragm has been proposed to precisely quantify the increased thickness of this muscle, together with an increase of the T2 signal and heterogeneity similar to limb muscles [190].
Not only can NMR serve for muscle imaging, but also for muscle spectroscopy, particularly of 31P. The few studies performed in GRMD dogs have highlighted similar abnormalities of the phosphorus spectrum as in DMD patients, including decreased phosphocreatine and increased phosphomonoesters and phosphodiesters. A splitting of the Pi was also seen in GRMD dogs, as a possible result of the presence of prenecrotic cells with increased membrane permeability [191, 192]. Interestingly these abnormalities of the phosphorus spectrum can be used as biomarkers for treatment effect, since they improved following gene therapy, consistently with MRI indices and overall muscle functional improvement [36, 39].
Ultrasound imaging: Despite a recent interest in muscle ultrasound for human NMDs, an easy access to such material, and the apparent simplicity of the technique, only one published study reported the use of this technique in dog models. XLMTM dogs underwent diaphragm dynamic ultrasound, revealing a thinner and more echogenic diaphragm in XLMTM and these abnormalities were reversed by MTM1 gene therapy [193].
Complementary evaluation methods.
Force measurement: Force measurement has been proposed as an evaluation method in dog models of NMDs, since it is frequently used in mouse models and in patients. Force measurements on isolated muscles have been performed in GRMD dogs, including eccentric contractions-induced force decrement studies, but such invasive procedure precludes longitudinal studies, and have thus been rarely used [194, 195].
The first non-invasive study of in vivo force measurement was performed in the GRMD dog model of DMD, and focused on the isometric force generated by the flexion or extension of the tarsus, evaluated longitudinally [196]. The measurement was performed on anesthetized dogs and required electrical nerve stimulation but no invasive procedure. The flexion and extension forces were decreased relative to healthy dogs, but only extensors deteriorated with age. Importantly this study showed a good agreement between force measurement performed in both hindlimbs, allowing for comparison between a treated limb and the contralateral untreated one as a control [196]. This has been done in several studies using local or locoregional cell or gene therapies, demonstrating a force improvement in the treated limb relative to the untreated one, in legs or forearms [35, 197]. This method is not only useful for the evaluation of local or loco-regional therapies, but has also proven useful to evaluate systemic effects of pharmacological approaches or myostatin reduction through crossbreeding [151, 198]. Aside from the muscle force per se, contractile properties have also been quantified, and the relaxation and contraction times were shown to be prolonged in GRMD dogs, and this could be improved pharmacologically [151, 196]. An eccentric contraction protocol has been setup in GRMD dogs, which evidenced a marked force decrement and thus provided a quantification of myofiber membrane susceptibility [199]. Altogether, these parameters obtained using in vivo force measurements have triggered an extensive and multi-faceted muscle function evaluation in the GRMD dog model.
Muscle force measurement was also used in the XLMTM dog model, using a device system adapted to the measurement of torque in dogs, inspired by the work on GRMD dogs, and now commercially available. XLMTM dogs present with a decreased torque generated by flexors of the tarsus, as well as a decreased twitch on tetanus ratio, and a right shifted torque-frequency relationship signaling the E-C coupling defect present in this disease [200]. All these abnormalities improved in the hindlimb, following locoregional gene therapy [37], and the torque also improved in both forelimbs and hindlimbs after systemic delivery of the AAV vector [38].
Joint angles measurement: In both DMD and dog models, contractures lead to abnormal joint angles and range of motion. In GRMD dogs, early studies focused on the tarsus joint angle at rest, measured using a simple goniometer, and showed that this angle was smaller in GRMD dogs, correlating with disease severity and hypertrophy of the Sartorius cranialis muscle [188]. A trend towards an improvement of this resting joint angle was associated with steroid treatment [198]. This method has then been extended to the measurement of resting, flexion and extension angles of the hindlimb joints (hip, stifle, tarsus), evidencing beneficial effect of a pharmacological therapy, and conversely a negative impact of myostatin reduction through crossbreeding on joint contractures [185, 189]. This method is highly accessible, simple to use even iteratively, non-invasive, and provides sensitive biomarkers. For these reasons, it should be considered more widely by teams working with DMD dog models.
Prospective cognitive evaluation: Aged dogs can spontaneously develop progressive degenerative, age-related cerebral changes associated with impairment of cognitive functions and for this reason, are also being recognized as relevant models for dementia of the Alzheimer’s type [201]. In some human NMDs, brain function disorders and cognitive impairments are widely described, in particular in DMD and ALS (reviewed in [202]), yet these features have only been studied on rodents so far [99]. Because of common selected genomic signatures including genes involved in neurological traits [95], dogs represent a relevant model in cognition, complementary to the rat in some instances. Cognitive tests have been developed and validated in canines, and their inclusion in preclinical trials would provide additional criteria for measuring treatment efficacy. A better understanding of how research dogs perceive their environment should improve welfare and keep the dog as a rationalized model.
Preclinical validation of gene therapies in NMDs
Following the success of gene therapy clinical trials relying on convincing preclinical studies in dogs [104, 204], gene therapy preclinical trials performed in dog models of NMDs have recently been a springboard for clinical trials.
Gene replacement with MTM1 and microdystrophin
An attractive way to treat monogenic genetic diseases is to provide a wild-type, healthy copy of the mutated gene. For the X-linked myotubular myopathy, the whole MTM1 cDNA is small enough (3.5 kb) to be packaged in an AAV vector. An AAV8 vector driving the canine MTM1 cDNA under the control of a muscle-specific human DESMIN promoter was produced [37]. Locoregional and systemic delivery of the AAV8-MTM1 vector was performed, and injected dogs benefited from a markedly and sustained functional improvement even when the treatment was initiated at a late stage of the disease [37, 139]. These convincing and highly promising results motivated the initiation of the ongoing ASPIRO phase 1/2 clinical trial conducted by Audentes Therapeutics. At 48 weeks post-injection in the first two cohorts of treated children, the well-tolerated gene therapy triggered robust tissue transduction, protein expression and significant improvement of motor and respiratory function with a majority of patients achieving ventilator independence [10], which was consistent with the preclinical data obtained in dogs.
Gene replacement in DMD is more challenging because of the large size of the DMD cDNA (14 kb) that precludes its packaging into an AAV vector. To overcome this size bottleneck, optimized microdystrophin cDNAs have been engineered, which retain essential functional parts of the coding sequence. Validity of the strategy in dystrophic dogs was initially obtained using intramuscular injections [114, 205]. Three independent groups working in parallel on GRMD dogs but using distinct AAV serotypes, promoters and microdystrophin constructs, provided evidence of the validity of a body-wide gene transfer using a systemic injection, yielding high expression levels of microdystrophin, absence of safety issue and substantial functional improvement [39, 206]. Hence, two of the three AAV-microdystrophin ongoing clinical trials launched in DMD boys rely on preclinical data obtained in GRMD dogs [207]. Optimization options include larger genes delivered through a dual AAV strategy. A first encouraging proof of concept study was recently performed in a GRMD dog, showing mini-dystrophin expression associated with functional recovery after intramuscular injection [208].
Exon skipping strategy for DMD
With the same objective to overcome the issue of the DMD cDNA size, exon skipping strategy has long been proposed, and consists in forcing skipping of one or several exons by the spliceosome machinery to restore the reading frame and produce a shorter, yet readable transcript. This can be achieved using antisense oligonucleotides (ASOs) complementary to splice sites, exonic splicing enhancers or exonic splicing boundaries, which are either vectorized by an AAV or used as naked sequences, with different chemically engineered structures. In GRMD dogs, two studies reported that skipping of exons 6 and 8 from the transcript using an AAV-U7snRNA strategy triggered high expression levels of a quasi-dystrophin and resulted in improvement in muscular function [35, 36]. Until now, clinical trials to assess the exon skipping strategy in DMD patients have focused on the use of ASOs, and mostly targeted exon 51 that is a hotspot of mutations in DMD patients. Eteplirsen, a phosphorodiamidate morpholino backbone of ASOs (PMO) targeting exon 51 has been the first drug approved for DMD by the FDA in 2016 [209]. Extensive preclinical studies focusing on antisense oligonucleotides have been performed in the CXMDj Japanese beagle strain, in which the GRMD mutation was introgressed [210]. Numerous studies, using PMOs, vivo PMOs (vPMOs) and more recently Peptide-conjugate PMOs (PPMOs) have investigated their therapeutic potential. In 2009, Yokota et al. reported for the first time a widespread dystrophin expression and overall functional improvement after iterative injections of a cocktail of PMOs inducing exons 6 and 8 skipping [18]. Then, new generations of PMOs with enhanced cell penetrability and improved pharmacokinetic profile have been tested. Indeed vPMOs were shown to be efficient in inducing prolonged dystrophin expression in comparison with PMOs [211]. Then the comparison was extended to PPMOs, which yielded low levels of dystrophin expression in skeletal muscles and no locomotor improvement, but heart expression of dystrophin associated with an improvement in cardiac conduction abnormalities in CXMDj dogs, attributed to amelioration of vacuole degeneration in Purkinje cells [165].
CRISPR/Cas9 genome edition strategy for DMD
In less than a decade, the repurposing of the CRISPR/Cas9 bacterial immune defense system into a highly efficient gene-editing tool has revolutionized therapeutic strategies to treat genetic diseases, including NMDs [212]. After successful studies in murine models of DMD, systemic AAV-driven therapeutic gene edition has been achieved in the ΔEx50 canine model of DMD harboring a mutation in the 5’ consensus donor splice site of intron 50 that induces skipping of exon 50 [47]. Using a guide RNA targeting the exon 51 splicing enhancer, the CRISPR/Cas9-mediated edition resulted in either reframing of exon 51 following a single base insertion, or in skipping of exon 51 and reframing between exons 49 and 52 [19]. Eight weeks after systemic delivery of AAV9 vectors in two one-month-old ΔEx50-MD pups, widespread dose-dependent expression levels of dystrophin were found in striated muscles, ranging from 3 to 92% of wild-type levels [19]. Although no functional characterization was provided in this initial report, these correction levels overcome the estimated threshold of 15% of normal dystrophin levels required to provide a significant therapeutic benefit for DMD patients [213]. Skipping several exons in some patients should be even more challenging, a condition shared with the GRMD dog. In this model, a CRISPR/Cas9-directed repair to correct the mutation has been tested, however yielding low levels of dystrophin expression [214]. Still, this new generation of gene therapy methods is emerging, and it is very likely that it will be a major component of the therapeutic toolbox to treat many NMDs in the future. Dog models will undoubtedly contribute to study immune response, off-target mutations, and long-term efficacy and safety of these advanced innovative therapies.
Phenotypic heterogeneity and translational precision medicine
Facing biological complexity, unraveling modifying genes
Two individuals carrying the same prevalent disease-causing genetic variant can express clinical signs with markedly different degrees of severity, a feature known as variable expressivity. This interindividual phenotypic heterogeneity is common in dogs and well exemplified in DMD models. In a single litter of GRMD dogs, some may display a severe phenotype with early loss of ambulation, while others will live with a very mild form of the disease, with all possible intermediates [215–217]. This biological evidence is shared with DMD patients who can exhibit a wide range of steroid responsiveness and motor, cardiac or cognitive alterations [218, 219]. However, even if interindividual variability [220] represents another similarity of canine models with their human counterpart, this has long been a source of criticism against dog studies, claiming that it prevents from quantifying significant effects of the assessed therapies. The many efforts undertaken to quantitatively describe the GRMD disease course, and to develop discriminating and sensitive evaluation methods as detailed above, have allowed for the clear demonstration that interindividual heterogeneity was manageable. Indeed, several studies in GRMD dogs succeeded in reliably concluding on the effect of a treatment, with a limited number of animals. For example, a therapeutic effect following pharmacological approaches could be detected in groups of six to ten GRMD dogs [151, 198]. This number of animals was even lower in more efficient gene therapy studies, for which smaller groups of three to five dogs allowed clear demonstration of a positive effect [35, 39].
Not only does interindividual heterogeneity better anticipate highly variable human conditions [218, 219], but it also represents an opportunity to identify disease modifiers in dogs carrying the same NMD-causing mutation and sharing the same environment and medical support. Indeed, the phenotypic diversity of GRMD dogs give rise to long survivors, mildly affected dogs expressing very discrete clinical signs. In the Brazilian GRMD colony, one sire and one of his sons were identified with such a mild phenotype. Linkage, genome wide association and muscle transcriptomic analyses of these two ‘escapers’ identified a heterozygous point mutation in the promoter of JAGGED-1, yielding overexpression of this Notch ligand [43]. This finding emphasized a putative role of the Notch pathway in DMD-like conditions, opening novel therapeutic targets. Also, PITPNA expression in muscles was markedly increased in severely affected GRMD dogs and decreased in the two escapers, compared to healthy dogs. Downregulation of PITPNA was shown to improve dystrophic muscle pathology in diverse models; it is associated with a decrease of PTEN and activation of the AKT pathway, counteracting the previously described dysregulation of this pathway in severely affected GRMD dogs [221, 222]. PITPNA downregulation in escapers thus strongly argues in favor of PITPNA as a DMD modifier and also a putative therapeutic target. Of note, the JAGGED-1 mutation has not yet been found in other DMD dog models, even mildly affected, strongly suggesting that new disease modifiers will spontaneously emerge from drifting DMD dog colonies worldwide. Indeed, comparison of mRNA and protein expression of muscles sampled in moderately and severely affected dogs from a US colony have led to the identification of new modifying candidates, such as the chitinase 3-like 1 gene (CHI3L1) found markedly overexpressed in severely affected dogs [223] and previously identified as a molecular component of the DMD muscle signature [224].
Another prominent example is the identification of a modifier gene in the ALS dog model. It was initially discovered in a panel of client-owned Pembroke Welsh Corgis that all carried two copies of the predisposing missense mutation in SOD1, but either expressed clinical signs at relatively early age (7–9 y) or reached 15 years of age without any signs of the disease. A genome-wide association study followed by a fine-mapping analysis using the two distinct clinical groups as cases and controls led to the identification of a 12.5-kb risk haplotype containing five SNPs in the coding and intronic regions of the SP110 gene, encoding for a nuclear protein. Presence of this haplotype was secondarily identified in other breeds to be predictive of developing degenerative myelopathy [79]. Besides, dogs with SOD1-related degenerative myelopathy support studies investigating pathogenic mechanisms involved in both canine neurodegenerative diseases and human ALS, thanks to a possible access to samples from the central nervous system of well clinically stratified dogs [160, 226].
Thus, dog models both help improve knowledge on severe disorders such as DMD or ALS, and contribute to elucidate the molecular pathways involved in neuronal and muscle homeostasis, offering novel targets for therapeutic strategies.
Model in precision medicine
Computer-assisted individualized decisions have already proved their efficiency in many steps of the medical path, including help in diagnosis, prognosis and therapeutic choices or dose scheduling through pharmacogenomics [227]. Because many of them receive extensive medical care, and display interbreed and within breed genetic variability [92, 228], dogs are relevant models for individualized consideration, an expanding field in veterinary medicine that is often connected with genetic analyses of dog’s history [229, 230].
Identifying biomarkers
DMD dog models have recently played a prominent role in finely describing tissue- and age-specific components of DMD pathogenic mechanisms. A panel of molecular and imaging techniques were used to characterize affected young and adult GRMD dogs, as well as specific signatures of the diseased cardiac versus skeletal muscle and of differentially affected skeletal muscles. In particular, transcriptomic analyses revealed downregulation of metabolic pathways in skeletal muscles, presumably contributing to the disease progression [231], and helped unravel differentially expressed proteins, such as the brain-derived neurotropic factor (BDNF) whose level is elevated in the cardiac muscle of young affected dogs [232]. From these analyses and others performed in GRMD dogs, biomarkers such as proteins or miRNAs in serum or urine have been identified, with a possible translation to the follow-up of DMD patients [232–235]. In this field as in many others, DMD dog models and other dog models of NMDs will likely promote the discovery of new, relevant, and minimally invasive disease biomarkers.
THE COMPANION DOG, A DEFENDED AND REVEALING ANIMAL
Each year, around 65,000 dogs are used for scientific purposes in the USA [236] and 18,000 in Europe, representing 0.16% of the total number of animals included in research protocols in Europe. The majority of these dogs (74%) are used in projects dedicated to research, development and quality control of products and devices for human medicine and dentistry and for veterinary medicine, toxicological and other safety evaluations. A small proportion (21%) is used for biological studies of a fundamental nature, including preclinical assays in the NMDs field [237]. These numbers are globally decreasing (up to 200,000 dogs were yearly used in the USA), and continuous progresses are being achieved to develop substitutive methods [238, 239], which will help optimize the overall number of animals used in research [240]. Yet, because alternative models do not still faithfully mimic the complexity of pathogenic mechanisms, regulations in drug development require preclinical trials in animals, and in most cases in at least two different species [241].
Every year, in many countries, media campaigns are organized to denounce the use of dogs for scientific purposes. One might think that this empathy for animals is a recent phenomenon, rooting in a consciousness awakened by the large panel of deleterious consequences of our modern way of living, which include climate change, biodiversity erosion, loss of food, water, air quality and animal abuse in some slaughterhouses. Of note, the debate over the use of animals for scientific purposes is an old one [242], and it is remarkable that the central question has never changed over the last two or three centuries, opposing two groups of people. On one hand, the advocates of the benefits to human health who legitimize the use of animals are facing, on the other hand, people who primarily consider the cost paid by animals and are thus defending the abolition of their use in research. An extensive study conducted in Denmark on a large panel showed that the first category of approvers represents 30 to 35% of people, while the second category of non-approvers represents 15 to 20% of people. These two categories represent half of the population. The other half is constituted by ambivalent, reserved people who indeed represent the complexity of the underlying ethical issue. For these people, the decision to approve or reject the research done on the animal must be taken on a case-by-case basis, weighing the animal cost and the benefit for humans [243]. This way of thinking is less polarized, and ultimately corresponds to the balanced regulations that most of the involved countries adopted. Improved animal welfare and reduced or absent pain is the central ethical question, which has for a long time been mistakenly obscured by a false debate opposing promoters or detractors of science [23].
The desire to recognize prevention or treatment of animal pain as a moral value has been expressed as early as the 17th century, but organization of militant associations happened only in the 19th century, during which experimental physiologists who were promoters of vivisections, have massively experimented on animals and especially on dogs, with almost no use of anesthetics or analgesics. Once discovered, these molecules have long been considered harmful for reliability of data gathered from experimental animals and seldom used. In response, the Society for the Prevention of Cruelty to Animals has been founded in London in 1822, followed by the creation of antivivisection societies in England, France and the USA between 1875 and 1883. These movements have heavily relied on the special status of the dog, who has become an emotional companion, recognized for his behavior reducing his distance from human, and has no longer been considered as an ordinary animal. This long-lasting activism led to voting the first laws to protect animals, in France in 1850, and in England in 1876 with the “Act to regulate vivisection”. Initial laws already aimed to replacing and reducing the number of animals used, but also to refining procedures performed by trained experimenters who obtained authorization after submitting their project to peers. These laws have since evolved, always reinforcing incentive towards a prevented or treated pain, and an improvement of animal welfare. Thanks to these regulations combined with openness on animal experiments, it is presently observed that many reserved people join approvers to form a large majority of citizens supporting a responsible research on animals [244].
Present ethical regulations
The use of animals for scientific purposes is strictly regulated. In France for example, according to the current European directive [241, 245], dogs have to be housed in groups with a minimum surface of 8 m2 for an adult golden or Labrador retriever, in annually evaluated licensed facilities. All the experimental protocols must conform to the 3Rs rule to be validated by an ethical committee and licensed by the competent authorities, before their initiation. In the case of invalidating diseases such as muscular dystrophy, the experimental procedures have to be classified as severe, meaning that a retrospective evaluation of the project has to be provided by the licensee. The experimenters must attend specific training and have to regularly update their knowledge and skills.
The evolving 3Rs guiding principles and humane endpoints in dog models of NMDs
The “3R” principle, which today governs ethics in animal research and in many countries has been integrated into the legislation [246], was first proposed by Russell and Burch in 1959 [247], based on the fact that “the humanest possible treatment of experimental animals, far from being an obstacle, is actually a prerequisite for successful animal experiments”. In this text, Russell and Burch considered that “inhumanity can be, and is being, diminished or removed under the three broad headings of Replacement, Reduction, and Refinement”. These 3Rs still remain cornerstones in animal research, and guide study conceptions to promote humane and rationalized treatment of the animals. More recently, a fourth “R” -for rehoming, has been added to the ethical guiding principles.
The application of the 3Rs rule in dog models of NMDs is facilitated by the special penultimate position of dogs in the translational process and by all the non-invasive evaluation tools available for these dogs.
Medical feedback for health and welfare of owner dogs
Human selection of desirable traits in dog breeds has been accompanied by the concomitant dissemination of morbidity alleles, often linked to the selected morphological traits [251]. As of July 2019, there are 409 deleterious inherited traits registered as models for human diseases in the Online Mendelian Inheritance in Animals database (OMIA, https://omia.org). As a result of this effort in comparative medical genetics, 290 variants in 190 canine genes have been identified. The majority (65%) was discovered in the last decade. Besides the obvious benefits for human medicine, the molecular identification of disease-causing variants has allowed the development of genetic tests that are now routinely used by breeders to avoid mating between sires and dams carrying deleterious alleles, mostly inherited as autosomal recessive traits. Indeed, it has recently been estimated that two out of five dogs from an international panel of 100,000 owners’ dogs carry one of the 152 deleterious known variants that had been tested [252]. Thanks to these tests and their assimilation by breeders who are now well informed of the disease-causing alleles that segregate in their breed [253], it has been shown from a panel of eight diseases that the disease-causing variant frequency declined by nearly 90% over 8–10 years [254]. Considering the very low number of research dogs used for initial genetic analyses, the benefit for the worldwide dog population is invaluable.
CONCLUSION AND PERSPECTIVES
Choosing relevant animal models in biological research implies recognizing properties shared with humans, based on the two biological concepts of unity across evolutionarily linked species, and diversity between species or between individuals [85]. For many reasons, the modern dog is likely the most appropriate preclinical model for neuromuscular diseases, cancers, eye, immune and neurodegenerative diseases, and behavioral disorders among others. The dog shares the longest common history with human, as well as his direct and daily environment. Spontaneously emerging disease-causing or -modulating variants segregate in the dog heterogeneous genome, a model anticipating precision medicine issues. Importantly, the long dog’s life expectancy, its large size and distribution volumes make it very comparable to that of human. Finally, its docility and cognitive performance allow to easily perform many non-invasive and non-sedated functional assessments.
However, for unanimously accepted ethical reasons, and also because of elevated costs, the use of research dogs raised in experimental colonies should be limited to studies that aim to answer the very last questions that cannot be faithfully addressed in routine laboratory animals, such as the mammalian rodents. One of these questions is the long-term preclinical assessment of efficiency and innocuity of innovative therapies in a heterogeneous panel of human-like individuals, mimicking more closely the “real life” clinical settings. In particular, it is mandatory for modern biological products that have recently been introduced to the market, for which data about unique or combined sequential therapies (AAV, ASO and RNAi for example) require to be expanded. This is of importance in the context of potential immune adverse reactions following repeat deliveries that may be necessary for some patients (Poster 147 in [255]). Of note, the number of research dogs could be further reduced by including affected pets in preclinical trials, with the consent of their owners. This has been previously envisaged for cancers [256] and would contribute to reward our loyal partner by improving not only its welfare through the generalized use of genetic tests, but also its health by facilitating its access to novel therapies.
If accepted, the benefit of including cohorts of affected dogs in the early phases of clinical trials may be hampered by the decreasing prevalence of morbidity alleles, counterselected by breeders using genetic tests. Presumably, the loss of relevant heterogeneous models with unique advantages should be anticipated by banking sperm from dogs carrying deleterious alleles, allowing further recovery of this invaluable genomic diversity.
In the future, many of the preliminary questions raised by the development of innovative therapies may be primarily addressed by alternative methods, combined with algorithm-based self-monitoring of connected patients [257, 258]. This will certainly reduce the pressure on the use of animal models in research, but likely will not spell the end of ethical issues that biomedical advances pose to our ever-changing societies.
AUTHORS CONTRIBUTION
IB and LT wrote the manuscript; CH performed bioinformatics analyses; LT conceived figures; all three authors edited and approved the manuscript
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
In the European Union, rarity is defined by a prevalence below one case in 2,000; in the US, when less than 200,000 people are affected.
ACKNOWLEDGMENTS
We thank Nicolas Van Caenegem, Ambre Jaraud, Lucie Chevallier, Stéphane Blot and Fanny Pilot-Storck for their help or comments, Eulalie Thiou for her artwork on ![]() , Nathalie Loux for providing information, Jordan Blondelle and Matthew Borok for edition of the manuscript, Fred Relaix for his efficient support and the three reviewers who helped us improve the manuscript with corrections and stimulating suggestions.
, Nathalie Loux for providing information, Jordan Blondelle and Matthew Borok for edition of the manuscript, Fred Relaix for his efficient support and the three reviewers who helped us improve the manuscript with corrections and stimulating suggestions.
We express our deep gratitude to the handful of pioneering admirable scientists who promoted the modern tools of canine genomics, an essential step in the increased knowledge on this animal, its history, evolution, physiology and diseases, and to the international community of researchers working on canine genetics and genomics. We are grateful to breed clubs, willing owners, breeders, health associations, veterinarians, responsible companies and national kennels that have worked together to identify variants and contributed to improving the health and well-being of dogs through the use of tests. We also thank zootechnicians and veterinarians who professionally work with research dogs and daily provide them with attention, care and consideration.
Our work is supported by the French Agence Nationale de la Recherche (ANR; CRBAnim infrastructure, ANR-11-INBS-0003), the CNRS, the Association Française contre les Myopathies (AFM-Téléthon; Translamuscle strategic pole), the Fondation maladies rares and the Fondation pour la Recherche Médicale.