
Abstract
Mutations in MFN2 cause a range of Charcot–Marie–Tooth disease (CMT) phenotypes with different inheritance patterns and underlying pathogenic mechanisms. Recently, a family with a dominantly inherited CMT harboring c.2222T>G (p.Leu741Trp) mutation in MFN2 has been reported for the first time. Here, we report a second family also with a dominantly inherited CMT harboring the same mutation, thereby confirming the pathogenicity of this mutation. Interestingly, the disease onset of this second family is much later than the previously reported cases.
Keywords
INTRODUCTION
Charcot–Marie–Tooth disease (CMT) is a group of clinically and genetically diverse hereditary disorders that predominantly affect peripheral nerves. CMT2, caused by abnormalities in the neurons or the axons, rather than the Schwann cells or the myelin sheath, is often referred to as axonal CMT. CMT2A is the most common CMT2 and can be caused by more than 100 different mutations in mitofusin 2 (MFN2) gene.
Interestingly, the clinical manifestations of MFN2 mutations are strikingly variable(1). A mutation in the MFN2 gene may have a normal phenotype, but it may also cause CMT in an autosomal dominant or autosomal recessive manner. The severity of the disease ranges from asymptomatic to severe early childhood-onset neuropathy, even in patients with mutations in the same protein domains. Some patients may have additional features, such as cognitive impairment, hearing loss or optic atrophy.
A family with a dominantly inherited CMT harboring c.2222T>G (p.Leu741Trp) mutation in MFN2 has recently been described for the first time. Four out of the nine affected family members were examined and sequenced. They had a clinical disease onset at around age 20, followed by slow disease progression(2). Here, we report a second family also with a dominantly inherited CMT harboring the same mutation, firmly establishing the pathogenicity of this mutation. However, the average symptom onset of this second family was at age 39, much later than the first family described.
PATIENTS
There were five affected members in this CMT2 family (Fig. 1). Genetic testing showed heterozygous MFN2 c.2222T>G (p.Leu741Trp) mutation in all five affected family members.
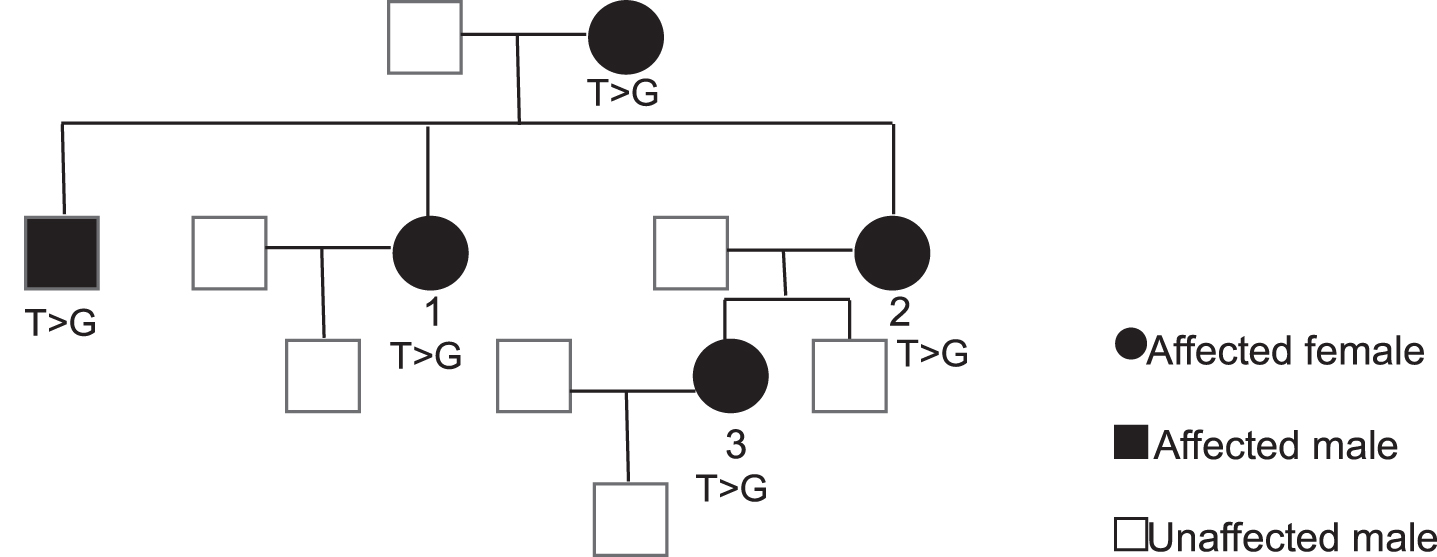
CMT family with MFN2 c.2222T>G (p.Leu741Trp) mutation. Pedigree shows the five affected member in this CMT2 family. Patient 1, 2, and 3 were seen and examined. The genetic testing revealed heterozygous MFN2 c.2222T>G (p.Leu741Trp) mutation in all five affected family members.
Patient 1
Patient 1 had chronic progressive hearing loss since her early 20’s but otherwise asymptomatic before age 49, at which point she started to experience numbness at the bottom of her feet. At age 61, the numbness and tingling has progressed to below the knees and in the fingers. She also had pain in feet and hands, mild problems with fine movement and leg weakness. Her exam was notable for mild pes cavus bilaterally, distal leg atrophy and tight Achilles tendons. She had normal strength in her arms and legs, except for 4 + (Medical Research Council score) in her first dorsal interosseous (FDI), abductor pollicis brevis (APB) and abductor digiti minimi (ADM) bilaterally, and 4 in anterior tibialis (AT) bilaterally. Sensation to pinprick was reduced at bilateral fingers and toes. Sensation to vibration was reduced at bilateral toes and knees. Prioprioception was intact at the joints, but Romberg sign was positive. She was diffusely areflexic and had abnormal toe, heel and tandem walk. Her nerve conduction study (NCS) revealed evidence of chronic mild-to-moderate, length-dependent, axonal sensory motor large fiber polyneuropathy, consistent with CMT Type 2. She scored 13/36 on CMT Neuropathy Score Version 2 (CMTNSv2)(3), which is in the moderate range (Table 1). Her genetic testing showed heterozygous MFN2 c.2222T>G (p.Leu741Trp) and heterozygous FIG4 c.2386C>T (p.Gln796*).
Scores of Patient 1, 2 and 3 using CMT Neuropathy Score Version 2(3)
Patient 2
Patient 2 had been clumsy since childhood. She developed numbness in the feet at age 40 and was diagnosed with CMT. At the time of our evaluation, she was 60 years old and had intermittent tingling, cramping, and pain in the legs; numbness to the knees and moderate difficulty buttoning shirt. Her other medical issues included mild hearing loss, hemiparesis of the diaphragm, heart murmurs, and alternating constipation and diarrhea. Her physical exam showed pes cavus bilaterally and tight Achilles tendons. There was atrophy of her hands and feet, as well as mild thinning in her legs. Her strength was 4 in bilateral FDI, APB and ADM; 4- in bilateral AT and left great toe dorsiflexion; and 0 in right great toe dorsiflexion and bilateral great toe plantarflexion. Sensation to pinprick was absent at bilateral toes and ankles. Sensation to vibration was absent at bilateral toes, and reduced at bilateral ankles and knees. Sensation to light touch and joint position was intact. Romberg sign was positive. She was diffusely areflexic. Mild tremor and mild scoliosis were observed. Toe, heel and tandem walk were abnormal. She scored 16/28 on CMTNSv2 (Table 1). Her genetic testing showed heterozygous MFN2 c.2222T>G (p.Leu741Trp) and heterozygous SH3TC2 c.1402_1403delinsTT (p.Ala468Phe).
Patient 3
Patient 3 first noticed numbness, tingling and pain in her hands at age 28. At the time of evaluation, she was 38 years old and back pain was the main issue. She had numbness up to below her ankles, but continued to run and stayed very active. Her physical exam was significant for high arches at the feet and atrophy of the feet and hands. She had mild weakness in hands: 4 + in FDI and ADM bilaterally. Her sensation to pinprick was absent at bilateral toes and ankles and reduced at bilateral knees. Her sensation to light touch and joint position was normal, but vibratory sense was reduced at the right toe and ankle. She was diffusely areflexic. She had slight difficulty walking with heels. Her NCS showed length-dependent, axonal sensory motor large fiber polyneuropathy. She scored 11/36 on CMTNSv2 (Table 1). Her genetic testing showed heterozygous MFN2 c.2222T>G (p.Leu741Trp), heterozygous SH3TC2 c.1402_1403delinsTT (p.Ala468Phe) and DNM2 duplication (Exons 1-2) with copy number of 3.
DISCUSSION
The family we described here is a second family with a dominantly inherited CMT2A harboring c.2222T>G (p.Leu741Trp) mutation in MFN2. Our report confirms the pathogenicity of this mutation. There are five affected members in this family, all of which were sequenced and three were examined. The clinical course is later in onset than the first family described. This is consistent with the literature on MFN2 mutations. The same mutation can be associated with different disease severity, even among members of the same family, which is probably due to environmental, epigenetic or other genetic factors that modulate clinical presentation.
Newly reported mutations are not only important for studying the pathogenic mechanism of CMT2, but also may have direct therapeutic implications. MFN2 is a mitochondrial fusion protein. Certain mutations (Arg364Trp, Leu76Pro) have been shown to have dominant positive effect in enhancing mitochondrial fusion(4), which in turn give rise to CMT2A phenotype. However, it is suspected that MFN2 c.2222T>G (p.Leu741Trp) mutation cause a loss of function in mitochondrial fusion(2). Agonists that could restore functions of defective MFN2 have been demonstrated in preclinical models. These agonists were found to be able to reverse mitochondrial defects in certain MFN2 mutations (Arg94Gln and Thr105Met) by promoting mitochondrial fusion and normalizing axonal mitochondrial trafficking(5). These MFN2 agonists could of interest for treating patients with this particular mutation.
All three cases in this study presented with varying degrees of pain in the hands and feet. It is therefore reasonable to suggest that pain is one of the phenotypes caused by the MFN2 variant. Pain has been reported in CMT caused by other mutations in MFN2, particularly in the late-onset form(1, 6). Common genetic variants have been shown to affect the development and perception of pain(7, 8). Rare, single-gene mutations causing painful phenotype are less common, but it has been shown in hereditary polyneuropathy(9, 10). The Leu741Trp variant of MFN2 would be adding to a growing number of single-gene mutations that can potentially lead to painful polyneuropathy.
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
None.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.