
Abstract
Dihydropyridine receptor congenital myopathy is a recently described congenital myopathy caused by dominant or recessive mutations in the CACNA1S gene. To date, only 11 cases from 7 families were described in a single report. Here, we describe a consanguineous family with three affected children, presenting congenital hypotonia, contractures, ophthalmoplegia and respiratory insufficiency, with a novel homozygous mutation in the CACNA1S gene. They also showed cognitive delay, pes equinovarus deformity and neurogenic changes that have not been associated with this myopathy in the previous reports. This report expands the phenotypic spectrum of dihydropyridine receptor congenital myopathy and underscores the importance of whole exome sequencing in early onset neuromuscular disorders.
INTRODUCTION
Congenital myopathies are a group of genetic muscle disorders, which are characterized on the basis of morphological features seen on muscle biopsy. There are early and late onset forms and the clinical course can be static or slowly progressive. Many of the congenital myopathies can be caused by mutations in more than one gene or mutations in the same gene can cause diverse muscle pathologies. On the other hand, the same mutation in a family can lead to different pathologic features [1]. Increasing use of exome and genome sequencing has led to identification of new congenital myopathy genes, but there are still many to be discovered.
Excitation-contraction coupling occurs at the triad. There is a specialized membrane structure, T-tubule and two sarcoplasmic reticulum saccules containing the ryanodine receptors. Dihydropyridine receptors are located on T-tubules. Activation of dihydropyridine receptors induce the opening of ryanodine receptors and release of Ca+2 from sarcoplasmic reticulum stores, which leads to muscle contraction [2]. The CACNA1S gene encodes the pore-forming subunit of the dihydropyridine receptor in the skeletal muscle. Heterozygous dominantly acting CACNA1S mutations have been associated with malignant hyperthermia susceptibility, hypokalemic periodic paralysis and thyrotoxic periodic paralysis [3–5]. Recently, both recessive and dominant CACNA1S mutations have been identified as a cause of congenital myopathy [6].
Here, we describe a consanguineous Turkish family whose three children presented with hypotonia, muscle weakness, respiratory distress, swallowing dysfunction, ophthalmoplegia and pes equinus deformity. Two of the siblings died at three months of age due to respiratory insufficiency. Whole exome sequencing showed a homozygous variant in CACNA1S in the surviving patient. The cases described in our report expand the spectrum of CACNA1S related congenital myopathy and show the importance of whole exome sequencing in diseases in particular associated with consanguineous marriages.
CLINICAL REPORT
The three probands are of Turkish descent and offspring of consanguineous parents (third cousin). In addition to the three probands, the couple had one healthy 10-year-old girl and one spontaneous pregnancy loss.
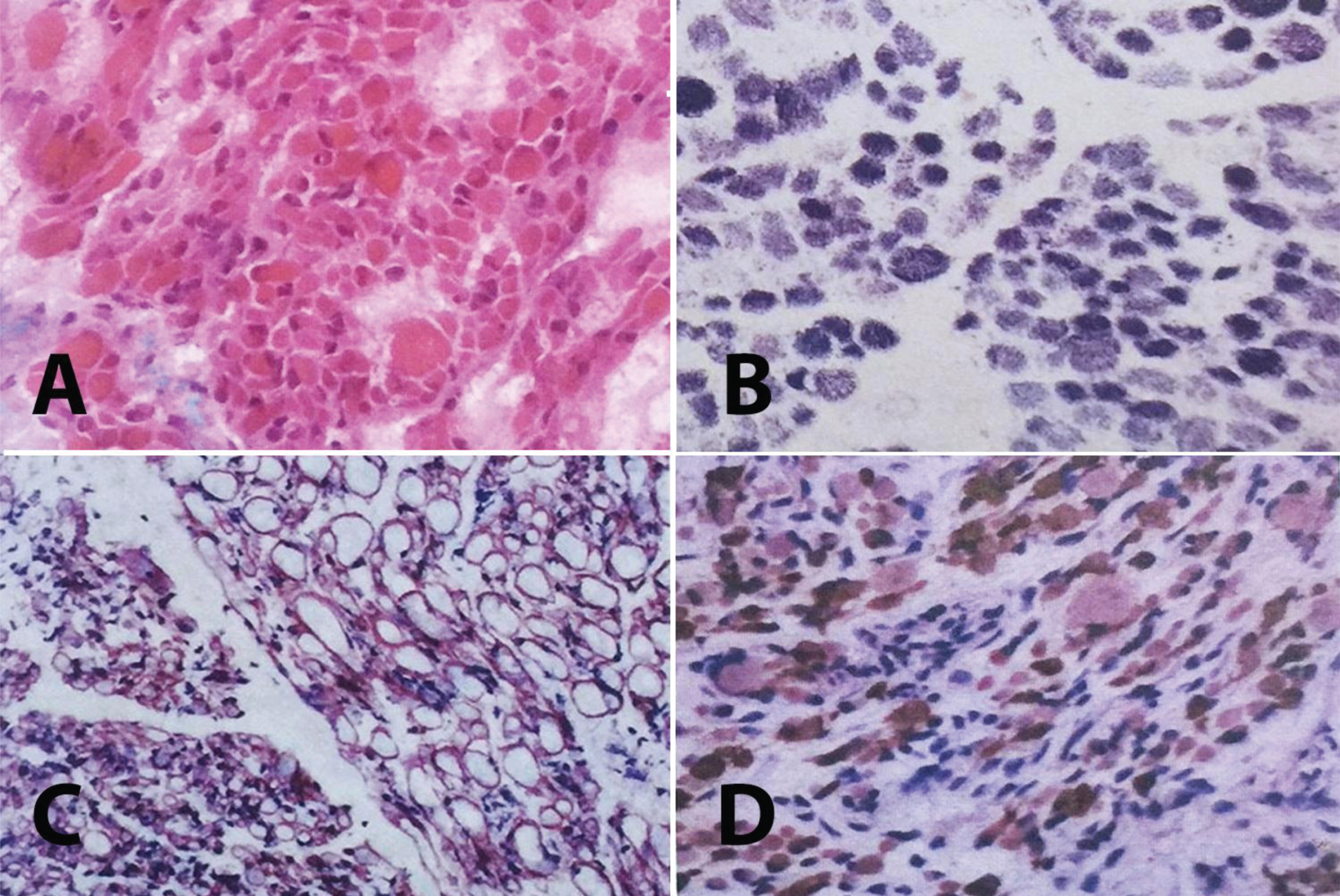
Muscle biopsy of Proband 2: A. Note the marked variation in fiber size and shape (HE×200). There were also hypercontracted fibers and increased fibrous tissue. B. There are no marked myofibrillary irregularities (NADH-TR×200), C. Normal sarcolemmal expression of merosin as well as grouping of large and small myofiber fascicles (DAB×200), D. Note the presence of huge type 1 fibers with fast myosin antibody (DAB×100).
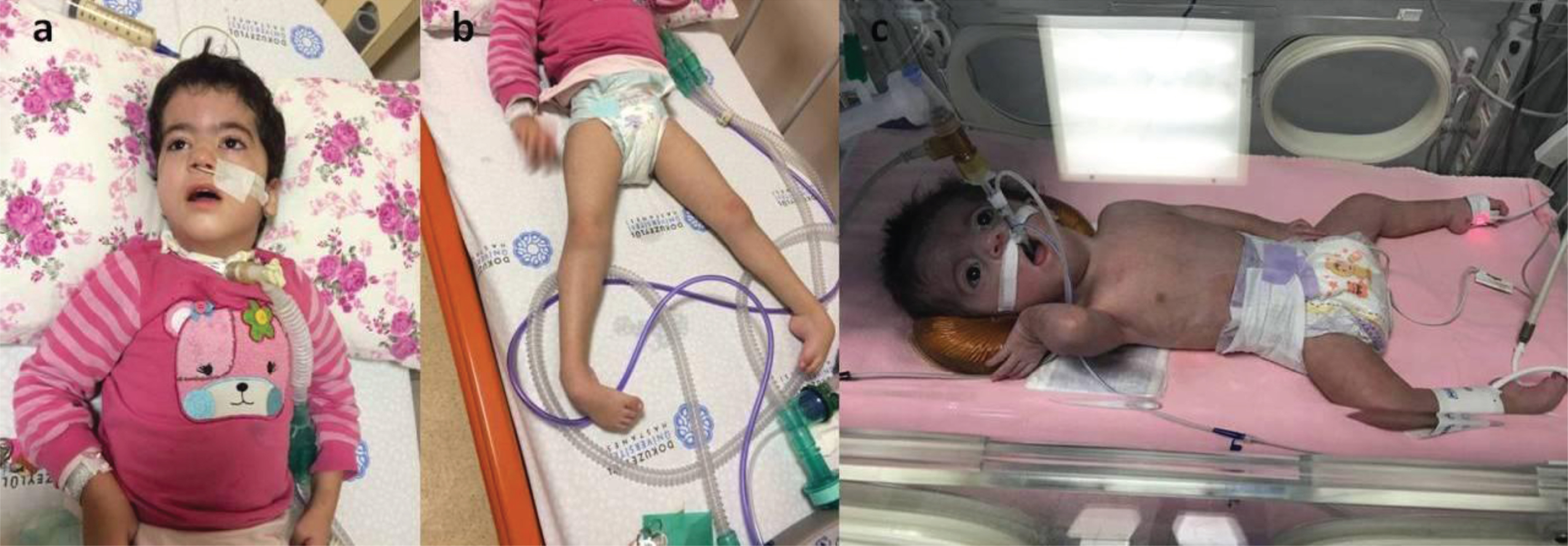
A-B: proband 2 with tracheostomy and nasogastric feeding. No anti-gravity movements are avaliable. She also has congenital onset pes equinus deformity. C: proband 3 has severe respiratory insufficiency. She also has congenital onset pes equinus deformity.
MATERIAL AND METHODS
Clinical studies
Probands 2 and 3, and their parents were enrolled in a research protocol for diseases related to consanguineous marriages that was approved by Dokuz Eylül University, School of Medicine Institutional Review Board as part of the Consequitur study. Informed consent, including for whole exome sequencing and the publication of medical information, was obtained from both parents prior to participation.
Whole exome sequencing and Sanger Sequencing
Whole exome sequencing (WES) was performed on DNA obtained from proband 2 and 3, and both parents, at the Broad Institute of MIT and Harvard, using Illumina Exome Capture Kit (38 Mb target). Sequencing data was analysed on the RD-Connect Genome-Phenome Analysis Platform (https://platform.rd-connect.eu/genomics) using standard filtering criteria for rare diseases, including Minor Allele Frequency (MAF)<0.01, Variant Effect Predictor (VEP) = mod/high and Combined Annotation Dependent Depletion (CADD) >20.
RESULTS
WES revealed a novel homozygous missense variant in the CACNA1S gene (NM_000069.3: c.2366G>A, dbSNP rs1157720606) that causes the substitution of a highly conserved (GERP = 3.91) arginine by histidine (p.R789H). Both parents were heterozygous for this variant. WES results and co-segregation of the CACNA1S variant with the disease in the family were confirmed by Sanger sequencing (Fig. 3a). No DNA was available for the other affected sibling with similar clinical findings. All bioinformatics prediction tools tested (Poylphen, SIFT, MutationTaster) classified the variant as pathogenic/damaging and the CADD score is 35. The variant was not observed amongst any of the 158,092 gnomAD exomes (allele frequency = 0) and was observed in heterozygous state in only one out of 31,382 gnomAD genomes (allele frequency = 0.000005278, https://gnomad.broadinstitute.org/variant/1-201038724-C-T, last accessed 6 April 2019). It is not reported in ClinVar, either. Moreover, the variant was not observed in a cohort of 1182 ethnically matched Turkish control exomes (TUBITAK-BILGEM).
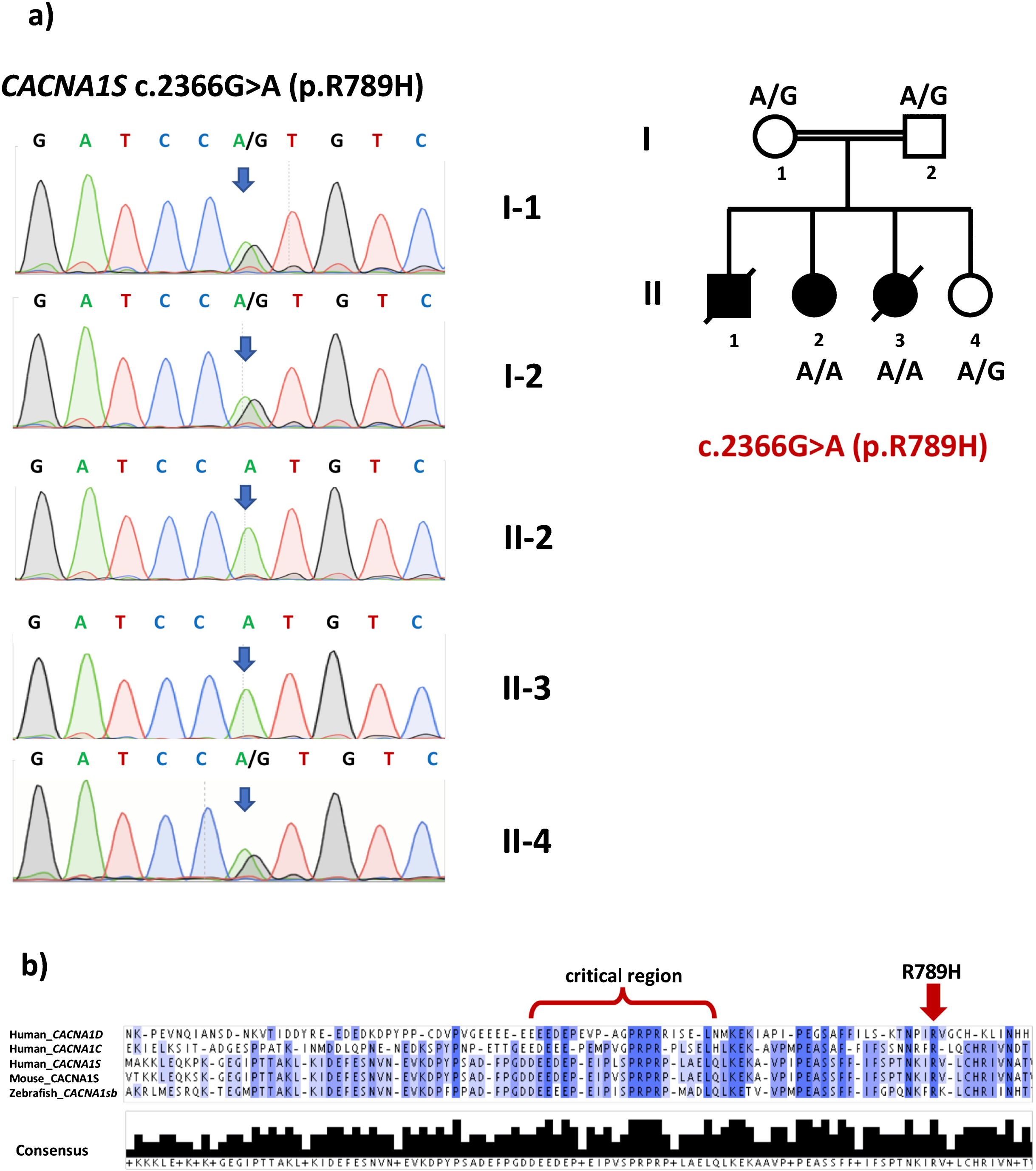
a) Pedigree and Sanger sequencing validation of WES results for CACNA1S c.2366G>A mutation. b) ClustalOmega multiple protein sequence alignment of the regions flanking the “critical region”, which is required for binding to Stac3, of human CACNA1S, human CACNA1C, human CACNA1D, mouse CACNA1S and zebrafish CACNA1Sb. Note that the region C-terminal to the critical region, that contains R789, is highly conserved. Darker colors indicate higher level of consensus between sequences. Lower panel indicates the level of consensus as a bar-graph.
DISCUSSION
Advances in genetic technologies have led to the identification of new congenital myopathy genes, most of which are associated with sarcomere structure and its stability. Despite this, about half of the patients with congenital myopathy remain without a genetic diagnosis. Very recently, mutations in the CACNA1S gene have been identified in 11 congenital myopathy patients from 7 families. Family history revealed recessive inheritance for two families and two families had dominant transmission. The cases were sporadic in the remaining three families. All patients in this cohort presented with early onset hypotonia and progressive muscle weakness with prominent axial involvement. All had mild facial involvement and four had ophthalmoplegia. Ophthalmoplegia occurred both in recessive and dominant cases. There were mild to severe respiratory and swallowing problems in all patients. There was no cardiac involvement and only two patients had elevated serum creatine kinase values. Six patients had scoliosis. All of the patients were still alive and their age ranged from 8 to 60 years [6]. Recessive cases were not different from dominant or sporadic cases. The clinical course in our patients were severe and only proband 2 is alive. She is now five years old. They had severe respiratory and swallowing problems and two of them died in the third month of life because of respiratory insufficiency. The surviving patient was dependent on mechanical ventilator all day and fed by percutaneous gastric tube. They all had ophthalmoplegia. The surviving patient also had scoliosis and lack of neck control showing the involvement of axial muscles. One striking finding was congenital pes equinus deformity, which was not reported in the previous cohort. The surviving patient also had moderate cognitive delay and ventricular enlargement with thin corpus callosum on brain magnetic resonance imaging, none of which were described previously. The cognitive status of other reported patients was not described. Cognitive delay and brain magnetic resonance imaging findings may be a feature of severe early onset cases but also may be due to prolonged neonatal intensive care stay, high oxygen exposure and episodes of respiratory insufficiency. Interestingly, muscle weakness and swallowing problems were less severe and there was no respiratory involvement in other reported recessive cases [6].
In the recent case series, histopathologic analyses of muscle biopsies showed an alveolar aspect of the intermyofibrillar network on NADH-TR staining, centralized nuclei, fiber size variability, core-like features, uniformity of type 1 and a dystrophic process. Ultrastructure of muscles on electron microscopy showed dilated T-tubules and sarcoplasmic reticulum and focal zones of myofibrillar disorganization. We could only evaluate the muscle biopsies of two siblings. Histopathological evaluation of the muscle biopsies revealed mild dystrophic and/ or myopathic changes, fiber size variability, nuclear internalization and fibrosis. In addition, many pathological immature myofibres were seen using neonatal myosin staining. Immunohistochemically, common structural proteins of muscle cell showed normal expression patterns and levels. All these changes except for the myofibrillary disorganization and vacuolization were similar to those previously reported [6]. Interestingly, there was also grouping of large and small size fibers, which is hallmark for denervation with reinnervation seen in neuropathies and spinal muscular atrophy. This finding was not reported before and no variants were found in hereditary neuropathy and lower motor neuron genes. On the other hand, overall early onset severe myopathy may present with type 1 predominance and can therefore have a neurogenic flavor; however, this may not be representative of an underlying neurogenic process, especially in the setting of a myopathic EMG and normal NCS.
Arginine residues in the S4 transmembrane helix of voltage-gated ion channels play critical roles in voltage sensing [7]. Indeed, most of the mutations identified so far in hypokalemic periodic paralysis 1 (HOKPP1) affect arginine residues within S4 voltage-sensing regions of Cav1.1, encoded by the CACNA1S gene, and most of them are arginine-to-histidine substitutions, just like the R789H variant that we identified in this study [8, 9]. However, localization of R789 to the cytoplasmic loop II-III of Cav1.1 points to a different role for this residue (Fig. 4). The II-III loop of Cav1.1 is critical for transmitting the excitation-contraction coupling to Ca2 + release by the gating of Ryanodine receptor (RyR1). This process is mediated by interaction of the loop II-III of Cav1.1 with the SH3-domain(s) of STAC3, STAC2 and STAC1 [10–12]. Recent studies identified residues 720–765, particularly 750–756, of Cav1.1 to be critical for binding to STAC3 [10, 11]. Localization of Cav1.1 to the cell membrane in muscle and neuronal cells depends on its interaction with STAC proteins, particularly with STAC3, and loss of these interactions lead to strongly reduced channel activity and perturbation of skeletal muscle excitation-contraction coupling [13]. Interaction of SH3-domain proteins with their ligands is mediated by proline-rich sequences flanked by an arginine on the ligand [14]. Yuen et al. identified R757 of Cav1.1 as a critical residue that makes multiple interactions via hydrogen bonding with the first SH3 domain of Stac2 [11]. Although the pathogenic variant R789H is not located exactly within the critical Stac3 interacting region, it sits in a highly conserved region (Fig. 3b) that is 25–30 amino acids apart from the critical region [15]. Moreover, this conserved region might possibly modulate the Cav1.1-Stac3 interaction [10].
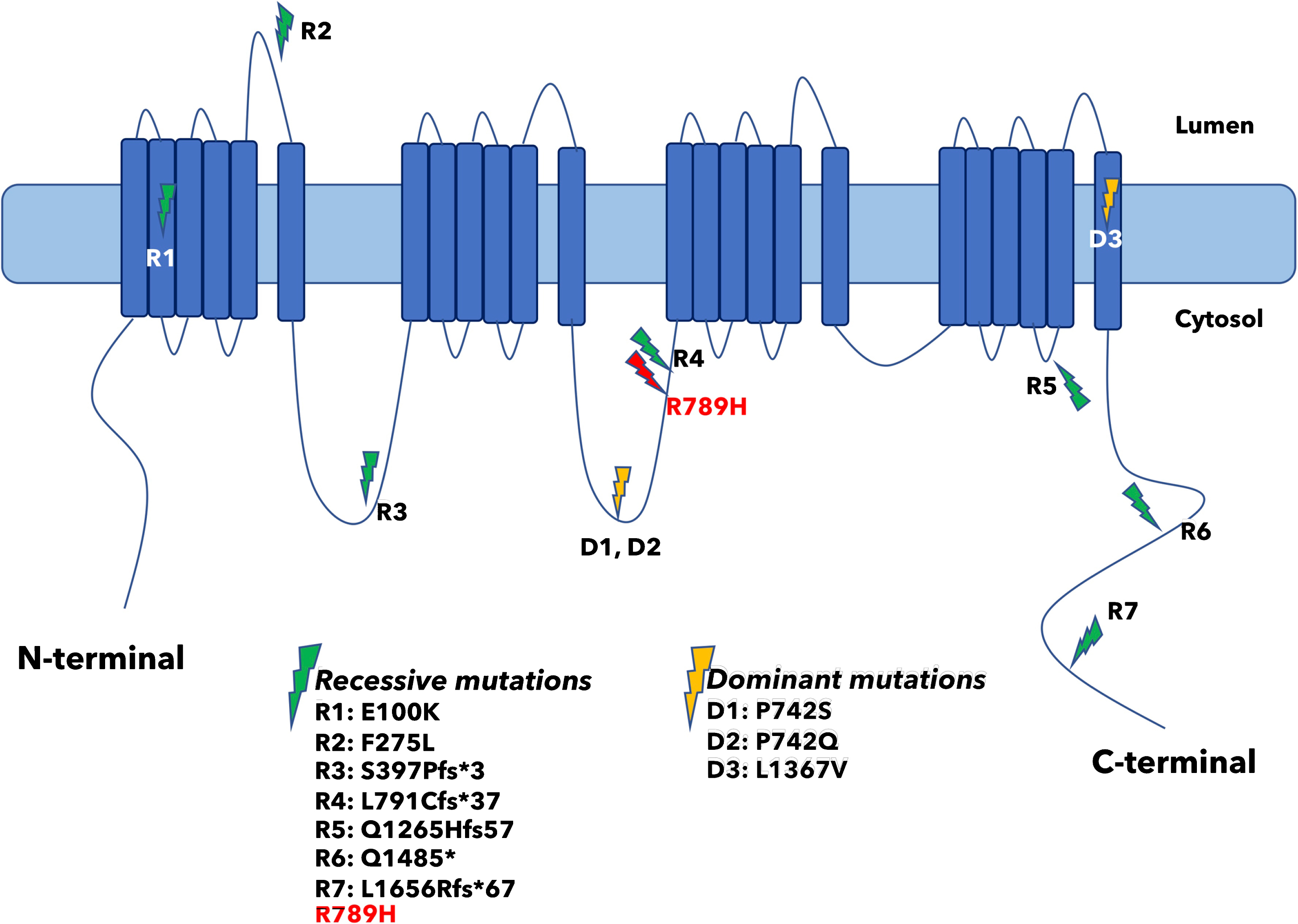
Localization of R789H mutation on the cytosolic loop II-III and distribution of previously reported CACNA1S/Cav1.1 mutations.
The severity of the phenotype observed in the three siblings presented here points to a critical role for the Arg-789 residue, and possibly the flanking region, in Cav1.1 functioning. Therefore, we propose that a possible molecular mechanism might be the perturbation of Cav1.1-Stac3 interaction, resulting in reduced channel activity and disruption of the excitation-contraction coupling. Interestingly, in two unrelated families reported by Schartner et al., the same residue P742, within the critical binding region to Stac3, was mutated to either glutamine or serine, with significant differences in the onset, severity and the range of tissues affected [6]. Further studies are required to provide mechanistic explanations to the observed discrepancies and to understand why R789H mutation causes such a severe phenotype.
In summary, the clinical course of CACNA1S congenital myopathy may be very severe and patients may die in the first months of life due to respiratory failure. Patients may also have cognitive delay and congenital pes equinus deformity. There may be neurogenic changes in muscle biopsy in addition to myopathic findings. Since clinical and histologic features of CACNA1S myopathy are diverse, this gene should be included in NGS panels of onset early onset neuromuscular disorders.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Footnotes
ACKNOWLEDGMENTS
This work has been supported by TUBITAK project 216S771 (UY, SH and YO). RH is a Wellcome Trust Investigator (109915/Z/15/Z), who receives support from the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), Medical Research Council (UK) (MR/N025431/1), the European Research Council (309548), the Wellcome Trust Pathfinder Scheme (201064/Z/16/Z) and the Newton Fund (UK/Turkey, MR/N027302/1). We thank the Broad Institute of MIT and Harvard for carrying out WES. The Broad Center for Mendelian Genomics (UM1 HG008900) is funded by the National Human Genome Research Institute with supplemental funding provided by the National Heart, Lung, and Blood Institute under the Trans-Omics for Precision Medicine (TOPMed) program and the National Eye Institute. We also thank the Centro Nacional de Análisis Genómico (CNAG), Barcelona, Spain for processing the raw data through the RD-Connect Genome-Phenome Analysis Platform (![]() ) developed under FP7/2007-2013 funded project (grant agreement n° 305444).
) developed under FP7/2007-2013 funded project (grant agreement n° 305444).