
Abstract
Background:
Genetic muscle disorders, including muscular dystrophies, congenital myopathies, and ion channel muscle diseases can be associated with significant disability.
Objective:
This study aimed to explore child and parent perspectives of the impact of living with a genetic muscle disorder.
Methods:
Eighty-three children (<16 years) with a clinical or molecular diagnosis were identified as part of a national prevalence study. Parents’ experiences and needs were assessed using a study-specific questionnaire. Additional outcome measures included parent and child self-report versions of the Behavior Assessment System for Children and the Pediatric Quality of Life Inventory. Parents also completed the Hospital Anxiety and Depression Scale and Activlim.
Results:
Sixty-four percent of families had a combined annual household income below $60,000 NZD ($43,650 USD), being less than the national median income of $73,000 NZD ($53,112 USD). Parents reported needing more support than they were currently receiving (40%), particularly with household chores (23%) and transportation (17%). Few parents (13%) or children (4%) reported significant child behavioral difficulties. Risks of impaired quality of life were high (parent proxy 71%, child report 70%), and associated with co-morbid health conditions (p = 0.008), functional status (p = 0.001), wheelchair use (p = 0.001) and mechanical ventilation (p = 0.01).
Conclusions:
Findings are relevant to those involved in the care and support of children, and their families, who are impacted by genetic muscle disorders. Targeted guidelines are required to inform the provision of services, alongside promotion of existing community services to improve access to financial support, and assistance with day-to-day functioning. Future research should examine intervention and treatment options aimed at maximising affected children’s quality of life.
INTRODUCTION
Inherited genetic muscle disorders, which primarily affect skeletal muscle, include the muscular dystrophies, congenital myopathies, and ion channel muscle disorders. Affected children have difficulties with motor skills due to muscle weakness, and are at risk of medical complications including cardiac, respiratory and orthopaedic difficulties. Some children have co-morbid cognitive disability [1]. There are no cures for genetic muscle disorders and while children are now living longer due to advances in supportive medical care [2], treatment continues to focus on symptom management, to optimise quality of life and functioning [3].
Health-related quality of life (HRQoL), defined as an individual’s perception of the impact of health and illness on the physical, mental and social aspects of their life [4], is increasingly recognised as a key outcome of health and rehabilitation services. Children with genetic muscle disorders have the potential to be at-risk of impaired HRQoL by virtue of living with a chronic and often progressive illness [5–7]. For example, Duchenne muscular dystrophy is associated with substantially impaired HRQoL compared to the general population [6, 8]. Yet, there has been little attention paid to HRQoL across the broader spectrum of genetic muscle disorders, despite variation in the muscles affected, severity, age of onset, and nature of progression. Improved understanding of experiences and needs across a range of conditions is needed to inform the development and delivery of services. Further, despite the early onset and medical impact of neuromuscular disorders, the behavioral and emotional profiles of affected children have received limited attention. Impact studies have tended to focus on physical symptoms in adults. The few paediatric studies are largely limited to parent-report and examine children recruited from single clinics with Duchenne muscular dystrophy [9, 10], which limits the extent to which findings can be extrapolated to children living with other neuromuscular disorders [11].
This study has three principal aims: to explore parents’ experiences of caring for an affected child; to examine the impact of these conditions on the HRQoL and behavioral adjustment of children using parent and child self-report; and finally, to identify factors associated with good or poor outcomes.
MATERIALS AND METHODS
Ethical approval
Approval for the study was obtained by the Health and Disability Ethics Committee of New Zealand (Reference number: 14/NTB/118) and the Auckland University of Technology Ethics Committee (Reference number: 14/296). All study processes comply with the Helsinki Declaration of 1975.
Study population
A large, population-based, epidemiological study of the prevalence and impact of genetic muscle disorders (MD-PREV study), sought to identify all living adults and children with genetic muscle disorders, residing in New Zealand (NZ) on 01 April 2015. Children with genetic muscle disorders in NZ are primarily cared for by a paediatrician with access to a paediatric neurologist. Based on a diagnostic classification outlined by Norwood and colleagues [12], genetic muscle disorders were defined as inherited disorders that primarily affect the skeletal muscles, encompassing both non-dystrophic congenital myopathies and muscular dystrophies as well as ion channel muscle diseases. Disorders of the anterior horn cell, neuromuscular junction and nerves were excluded. Multiple and overlapping sources of case ascertainment were used, including medical record searches tailored to each District Health Board in NZ, using combinations of keywords and/or International Classification of Diseases and Related Health Problems (ICD-10) codes. Similar search strategies were used to check NZ Ministry of Health records, the NZ Neuromuscular Disease Registry, and Genetics Service databases. Advertisements to encourage self-referrals to the study and contact with relevant community support organisations also aided case ascertainment. For all potentially eligible symptomatic and asymptomatic cases, medical records (including investigations and test results) were obtained to confirm details of each diagnosis. Study eligibility was confirmed by a neurologist. Cases with insufficient evidence to confirm a diagnosis were excluded.
For inclusion in this sub-study, children (aged <16 years at the point prevalence date) needed to have clinical or molecular confirmation of muscular dystrophy (including Duchenne, Becker, limb-girdle, facioscapulohumeral, Emery-Dreifuss, myotonic dystrophy or congenital muscular dystrophy), congenital myopathy, or ion channel muscle diseases (i.e. myotonia congenita or periodic paralysis). The parents of affected children were contacted and invited to complete an impact assessment. Written informed consent was obtained from all parents. Children were also invited to participate and provided written assent where deemed appropriate.
Study procedure
Those consenting to participation completed an impact assessment in-person with a trained researcher (66.3%, 55/83) or by mail (27.7%, 23/83) or online (6.0%, 5/83). Parents and children completed age-appropriate versions of the Behavioral Assessment System for Children-Second Edition (BASC-2) to assess child (2.6–18.0 years) behavior in the home and community. Core domains included externalizing behavior (e.g. hyperactivity, aggression), internalizing behavior (e.g. anxiety, depression), adaptive (prosocial) skills (e.g. social skills, leadership skills), and a behavioral symptoms index that assessed overall behavioral problems. Using linear T scores that are scaled with a predetermined mean of 50 and a standard deviation of 10, each parent-report subscale and composite measures were examined, along with a corresponding child-report when the same subscales were available. Across all subscales and based on recomendations from the creators of the measure, scores >69 were taken to signify clinically significant maladjustment, with the exception of the adaptive subscale where a cut-off score ≤30 indicates significant difficulties [13].
HRQoL was assessed using the parent and child (8.0–12.0 years) report versions of the 23-item Pediatric Quality of Life with Generic Core Scales (PedsQLTM GCS) version 4.0. The PedsQL GCS assess Physical functioning (8 items), Emotional functioning (5 items), Social functioning (5 items), and School functioning (5 items). Average emotional, social, and school functioning was captured in a psychosocial health score, and the total PedsQL score was an average of four subdomain scales. Items were scored using a 5-point Likert scale to reflect difficulties with each item, ranging from 0 = never to 4 = almost always. Example items included ‘Feeling angry’ (emotional function) and ‘Paying attention in class’ (school function). In accordance with standard scoring instructions, each item, including reverse scoring, was rescaled on a 0 to 100 scale (0 = 100, 1 = 75, 2 = 50, 3 = 25 and 4 = 0). Published cut-off scores were applied for both child self-report and parent proxy-report. One standard deviation below the mean of the population sample indicates at-risk status for impaired HRQoL [14]. Higher scores indicate better HRQoL.
A specific 25-item module for neuromuscular disorders, the PedsQLTM Neuromuscular Model (PedsQLTM NMM) [15] version 3.0 was also administered. This module consists of three scales: About my child’s neuromuscular disease (symptoms/function, 17 items), Communication (3 items), and About our family resources (5 items). The NMM has been validated in Duchenne muscular dystrophy and spinal muscular atrophy populations [15]. The instructions, response and scoring methods were the same for both Peds QL modules. Higher scores indicate better HRQoL.
Study specific questionaires were used to capture the following: demographic characteristics (child age at point prevalence date, gender, ethnicity); health information (diagnosis, molecular confirmation of diagnosis, need for ventilation and wheelchair use); and environment factors (estimated household annual income, marital status, and the responding parents’ mood and employment status). To determine the presence of any co-morbid health conditions, parents were also asked “Does your child have any other medical conditions?” Parent anxiety and depression were assessed using the 14-item Hospital Anxiety and Depression scale [16]. This standardised measure is a psychological screening tool that assesses symptom severity and caseness of anxiety disorders and depression in patients with illness and the general population. Total anxiety and depression scores range from 0–21 (normal 0–7; mild 8–10; moderate 11–14; severe 15–21). Child functional impairment in terms of activity functioning was determined using the ACTIVLIM [17].
Statistical analysis
A sensitivity analysis compared the characteristics of those families who were included in the current analysis (N = 83) and those families who were not (N = 76). T-tests for continuous variables and chi-square tests for categorical variables were used. Descriptive statistics were used to report frequencies, percentages, means, and standard deviations (SDs). Factors associated with poor outcomes were identified using t-tests to compare scores between those with and without each characteristic of interest. Missing data were managed using case or listwise deletion depending on the pattern of missing data in each analysis. Statistical signifcance was determined at the p = 0.05 level. All data analysis was performed using IBM SPSS software, version 23.
RESULTS
Sample characteristics
The prevalence study identified 159 affected children living in NZ on the point prevalence date. Children had a mean age of 9.04±3.75 years [range 0–15 years], with the mean age of parent-reported symptom onset being 2.01±2.67 years [range 0–11 years]. The majority of children were male (73.5%) and NZ European (77.1%, higher than the national average of 67.3% for children aged 0–14 years [18]). Duchenne muscular dystrophy (n = 61, 38.3%) and congenital myopathy (n = 38, 23.9%) were the most common diagnoses. Of the 122 families contacted, 83 (68.03%) parents (one parent from each participating family) and 61 affected children completed an impact assessment (Fig. 1). The non-enrolled sample were similar to those families included in the impact analysis (52% of the prevalence sample) on all characteristics shown in Table 1 (p > 0.05). Due to the search strategy, most cases were initially identified from the NZ Neuromuscular Disease Registry [19], followed by hospitals/neurologists and genetic services. The majority of parent respondents were mothers (88.0%, 73/83), who were married (81.5%, 68/83). More than one third of children (38.6%, 32/83) had some form of parent-reported co-morbid health condition (i.e. attention deficit hyperactivity disorder, learning delays, autism, cerebral palsy, anxiety disorder). Half of parents (50.6%, 42/83) had health conditions themselves, most commonly anxiety and depression, or physical ailments (e.g. back, neck, and knee complaints), followed by muscular dystrophy, fatigue, or other medical conditions (e.g. gout, headaches).
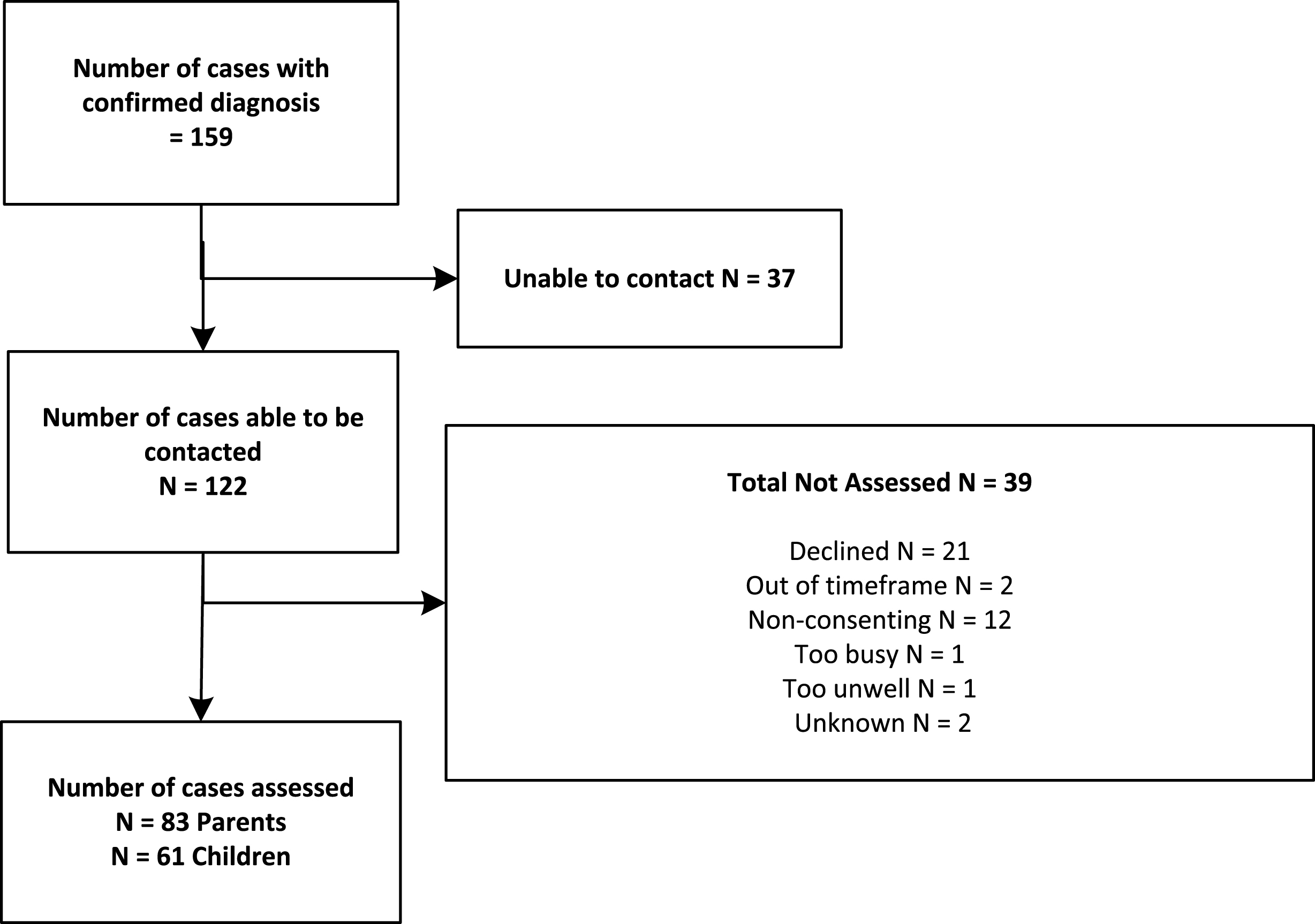
Flowchart of parent and child recruitment.
Sample characteristics
†N = 51 due to 32 cases missing or non-symptomatic. *N = 80 due to missing data. ±N = 81 due to 2 cases missing data. ‡Income data were available for 77 families due to missing data. ≠ Time off work due to child’s health or medical care.
Parent’s experiences, needs, and mental health
The majority of parents were ‘very’ to ‘mostly satisfied’ with the health care of their affected child (Table 2). Just under a third (31.3%) had received some form of unpaid support from family or friends. Nearly 40% reported unmet needs, and felt they needed additional help in caring for their affected child, including help with household chores (22.9%), transportation (16.9%), and financial advice (15.7%). While 89% of families were receiving some form of financial benefit (e.g. disability allowance), only 34% reported receiving any paid carer support, such as a person coming into the family home each week. 24.1% of parents reported unmet needs with regard to financial assistance, with the majority working only part-time (36.1%) or not at all (33.7%) (Table 1). Over half (63.8%) of families had a combined annual household income of less than $60,000 NZD ($43,650 USD), which is below the national median of $73,000 NZD ($53,112 USD) [20]. A third of parents reported paying some form of treatment-related costs for their child, including medications (i.e. Deflazacort, Melatonin), nutritional supplements (i.e. creatine, multivitamins), and complementary therapies that were not funded by the Government at the time of the study. Mean parent-reported anxiety and depression scores were within the normal range, with 13% reporting moderate to severe anxiety. Less than 3% of parents reported moderate to severe depression.
Parent-reported satisfaction, support, and needs
*Figures total more than 100% due to multiple responses from participants. Note: HADS Depression – I x item 9 imputed (all other items = 0), 1 x item 1a, 10d, 11a imputed with mean score = 1.
Child behavior and HRQoL
As a group, mean parent proxy and child self-report ratings of behavioral functioning using the BASC-2 ranged from 41.84 to 57.39, and were within the normal range for all subscales [13] (Table 3). Few children and parents reported clinically significant behavioral problems, being those difficulties likely to create serious problems in life (parent-report 4.8 to 13.3%, child-report 1.9 to 3.8%) (Table 4). Children reported fewer behavioral problems than their parents.
Means and SDs on parent and child reported BASC-2 and PedsQL items
Abbreviations: M = mean; SD = standard deviation; BASC = Behavioral Assessment System for Children; PEDSQL = Pediatric Quality of Life Inventory. *N = 62 due to missing data because of measurement age restrictions. †N = 71 due to missing data. ≠N = 40 due to missing data. ≠N = 39 due to missing data. μN = 23 due to missing data. Dashes (–) denote data not available.
In contrast, mean parent proxy and child self-report HRQoL scores using the PedsQL Generic Score Scales, ranged from 41.04 to 67.34, and were within the at-risk range for physical, social, psychosocial, and total HRQoL. Mean child self-report scores for school function were within the at-risk range, while mean parent proxy-report scores were not. Overall, children reported higher mean levels of HRQoL compared to parent proxy-report, with the exception of school functioning (parent-proxy mean = 59.35 (SD = 21.45), child-report mean = 57.91 (SD = 21.55). Mean parent proxy and child self-report scores using the Peds QL Neuromuscular Model ranged from 57.55 to 72.83.
In terms of the proportion of children with clinically significant problems, the PedsQL Generic Score Scale total score revealed a high percentage of children at risk of impaired HRQoL (parent-proxy 71.1%, child self-report 80.0% (Table 4). Thirty-two out of 46 children agreed with their parent’s rating of their at-risk status for HRQoL (69.5%). When compared to a healthy population, children were more likely than their parents to report risk of impaired HRQoL.
Proportion of children with difficulties in each domain
†N = 71 due to missing data. *N = 39 due to missing data. ‡N = 40 due to missing data. Dashes (–) denote data not available. PedsQL Generic score cut-off points were <65.4 for parents and <69.71 for children.
Factors associated with parent– reported child outcomes
Subsequent ANOVA analyses compared associations between types of diagnosis (Becker, Duchenne muscular dystrophy, congenital muscular dystrophy, congenital myopathy, and other diagnoses) and parent proxy-report outcomes. No significant group differences were found for behavior (externalising F(5, 74) = 0.86, p = 0.50, internalising F(5, 70) = 1.33, p = 0.26), total HRQoL, (F(4, 74) = 2.04, p = 0.09), parent anxiety (F(4, 73) = 0.44, p = 0.77), or depression F(4, 73) = 1.78, p = 0.14).
Factors associated with child HRQoL and behavior
Based on parent proxy-report, measures associated with greater disability, specifically poor functional status, co-morbid medical conditions, in-home mechanical ventilation, and wheelchair use were significantly associated with risk of impaired HRQoL. At-risk status for impaired HRQoL was significantly more likely to be reported by those parenting alone. Externalising and internalising child behavior problems were associated with parenting alone and lower family income.
DISCUSSION
This nationwide, population-based study of children living with a genetic muscle disorder examines parental experiences, and both parent and child perceptions of child behavior and HRQoL. Study strengths include: the multi-domain examination of child development beyond the usual primary focus on physical function; the use of outcome measures previously validated in pediatric neuromuscular disease populations, enabling international comparison; and the inclusion of a broad range of diagnoses to facilitate exploration of some of the less commonly studied, rare muscle diseases.
In common with previous studies [21–23], we found that parents were mostly satisfied with the medical care of their child. The financial impact of caring for a child with a genetic muscle disorder is a key concern for parents. The overall employment rate for predominantly mothers in our sample is similar to rates for partnered and solo mothers in NZ [24]. However, while income data were not normalised for the number of individuals in the household, just over one quarter reported that their financial situation had deteriorated since their child become symptomatic. Nearly 20% reported taking time off work in the past two weeks due to their child’s health and/or medical appointments. The extent to which parents’ employment is impacted may be even higher, given the impact on employment was not recorded for both parents, where relevant, in the current study. These findings align with prior evidence suggesting that as many as 50% of parents reduce their working hours, and/or have high levels of absenteeism to find the time to care for their affected child [25]. Further, the combined stress of working outside the home, whilst parenting a child with a disability, may manifest as parental ill health. Half of all respondents in our study reported personal health issues, including physical ailments, headaches, and fatigue. This rate appears to be higher than those reported based on normative samples. For example, a survey of a nationally representative sample of mothers in the United States (n = 8,060) found 7% reported fair to poor health [26].
However, there was also evidence of parental resilience. Mean HADS anxiety and depression scores, and the rates of moderate to severe anxiety in our sample revealed comparable levels to published normative samples of males and females, across a similar age range [27]. In fact, rates of moderate to severe depression, in our sample, were lower than found in normative samples. Given the lifelong nature of many genetic muscle disorders, associated stressors may become the new ‘norm’ for affected families. These findings are also consistent with prior suggestions that parenting stress in this population is related to child behavior rather than the genetic condition or specific disability [28].
While 89% of families reported receiving some form of government financial assistance, many families appear to be alone in their efforts to meet the daily care requirements of a child with a genetic muscle disorder. Some families may not see the need for additional support, however, these findings suggest that some families with an affected child are either not aware of, or do not qualify for government-level support that enables access to paid carer assistance. Alongside emerging evidence of significant caregiver burden in families with a child affected by genetic muscle disorders [29], the identification of a potential lack of awareness of available supports, suggests that it is important to improve the provision of supports to affected families. Landfelt and colleagues recommend a holistic approach to service provision and follow-up, including supporting families with activities of daily living, equipment and medical needs, and monitoring for caregiver well-being [29].
The findings of this study suggest that a high proportion of affected children are at-risk of impaired HRQoL. Parent proxy-reports of impaired HRQoL for children with Duchenne muscular dystrophy are common, especially in the physical domains [11]. However, few studies have examined a broad range of genetic muscle disorders, and the value of including child self-report has often been overlooked. In a review of 19 studies of HRQoL in paediatric Duchenne muscular dystrophy populations [11], only six studies had included parent and child (son) report, with poor to moderate concordance between raters. While our findings show that parents and children tended to agree on ‘at-risk’ status for HRQoL, children were more likely than their parents to reflect risk of impaired HRQoL compared to a healthy population sample. However, when mean child and parent proxy-report are compared in the current study, children’s mean HRQoL and behavior scores tended to reflect better outcomes than the ratings provided by their parents. In other words, children reported better HRQoL outcomes than parents but poorer overall outcomes when compared to the mean ratings of healthy children.
Similar findings have been observed across a number of studies examining children with chronic illnesses [6, 30–32]. Landfeldt and colleagues (2015) examined HRQoL in Duchenne muscular dystrophy, interviewing 770 parent-child pairs across four countries [6]. Findings revealed that children predominantly reported better outcomes than their parents. There are a number of possible explanations for parents overreporting the extent of impairment in their children’s HRQoL. It has been suggested that children may have adapted to their illness better than their parents [33]. Parents may experience feelings of guilt around the genetic aspect of their child’s condition, and their personal worries about their child and awareness of the disease trajectory may cloud their judgement of the impact of illness on their child. In contrast, children were more likely than their parents to report difficulties at school, including problems paying attention, remembering things, keeping up with school work, and missing school for health-related reasons. This is an area that may require further attention to ensure children’s academic, social, and behavioral needs at school are being met.
Consistent with limited previous research [34, 35], associations were found between risk of impaired HRQoL, and measures reflective of greater disability, including poor functional status, in-home mechanical ventilation, and wheelchair use. A recent systematic review, including 38 studies, found disease-related factors have a negative impact on HRQoL in individuals with genetic conditions [34]. In a parent-reported HRQoL study of 109 children with neuromuscular conditions, children on home mechanical ventilation had significantly lower mean total PedsQL scores than non-ventilated children [35]. Similarly, associations between wheelchair use and poorer HRQoL have previously been reported. A study of 99 boys (mean age 10.7 years) with Duchenne muscular dystrophy found consistent associations between fulltime use of wheelchairs and worse HRQoL Generic Core total and Generic Core physical summary scores based on both parent proxy and child- self-report [33]. Rather than being a direct result of ventalilation or wheelchair use per se, children with poor respiratory health and physical mobility problems may face additional challenges in their daily lives due to advanced disease progression, and may be at greater risk of social isolation. This premise is supported by evidence in the current study of links between impaired HRQoL and co-morbid medical complications. These links are likely the consequence of a more severe disease process and muscle degeneration, that may be associated with reduced capacity and opportunities for children to participate in a variety of physical and social activities outside of the home or at school.
In the current sample, few children were characterised by significant behavioral adjustment problems. This finding was somewhat unexpected given evidence of psychosocial and behavioral challenges for boys with Duchenne muscular dystrophy [36]. However, as previously noted, only a subset of boys with Duchenne muscular dystrophy are affected by such difficulties [37]. Furthermore, a review of parent ratings of 86 children with Duchenne muscular dystrophy, and their unaffected siblings, found no significant differences in behavioral adjustment in 80% of cases [38]. While current findings also found no significant differences in child outcomes and parent mental health between types of diagnoses, small sample sizes for each disorder may have affected this part of the analysis. Nevertheless, our findings attest to affected children representing a resilient and clinically heterogeneous group, many of whom continue to appropriately manage their behavior despite living with associated physical challenges. However, given the progressive nature of many of these disorders and the potential impact of pharmacological treatments on child development, behavior and mood [39, 40], ongoing evaluation and monitoring is recommended.
Significant child behavior problems in this study tended to be reported by families with solo parents (i.e. never married, divorced, seperated, or widowed), and those with limited financial resources. Socio-economic status has long been recognised as a mediator of developmental outcomes in normative samples. According to the family stress model, financial pressures exacerbate emotional and behavioral challenges for parents, such that they adversely impact on parenting and children’s outcomes [41]. Together, these findings suggest that parents who are facing financial hardship, whilst attempting to deal with the emotional and physical stressors of raising a child with a genetic muscle disorder as a solo parent, are a group who are likely to benefit from additional support.
There are some study limitations to acknowledge. As with any prevalence study, it is possible that some cases were not located, and this may have introduced bias to the study sample. Sampling bias is also possible, given those families who were most stressed, not coping, and/or in the greatest need of additional support, may have been less inclined to participate in an impact assessment. Whilst families who feel they are coping well, and are less stressed, may be more likely to have time and energy available to support study participation. In terms of HRQoL, future studies should also consider associations with psychosocial factors, such as child coping strategies, illness perceptions and self-esteem. Such research could reveal additional avenues for supporting children and minimising the adverse impacts of living with a genetic muscle disorder. Further, it must be acknowledged that not all paediatric neuromuscular conditions were included in the current study. There is overlap between primary myopathic conditions and other non-myopathic neuromuscular conditions as regards clinical phenotype and psychosocial consequences. However, these are clear neuro-anatomically distinct disease groups. These distinctions, along with feasibility and financial constraints led to the decision to focus on purely genetic myopathies. The nature of de-identified data in the current study did not allow exploration of the potentially differential impact for families with one child affected compared to families with more than one affected individual. It is also important to recognise that the significance of associations found between demographic, health and environmental factors and child outcomes may be minimal if examined in a larger sample using regression modelling. Nonetheless, this study examined a population-based sample representative of children and parents living in urban and rural areas, including those not currently in regular contact with health care providers. This has resulted in the examination of a large sample of parents, and their children living with genetic muscle disorders including those with poorly understood rare conditions. Added strengths are the inclusion of parent and child self-report data across multiple areas of functioning, and the identification of factors associated with outcomes that warrant further attention.
In conclusion, key findings of this study were the identification of significant deficits in the level of financial and in-home support provided to families, to assist with day-to-day provision of care for their child. Our study found a significant financial impact on families, with combined household income commonly below the national median. Service delivery needs to target these areas, assisting families to access services, and improve awareness of existing community support services. Clincians, educators, and parents need to be aware of the increased risks for children of poor HRQoL, with children in particular identifying difficulties at school. Future research is required to identify opportunities to promote better HRQoL for children with a genetic muscle disorder, with emphasis on early intervention to minimise the potential impact for affected children and their families.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
We are indebted to the participating families and their children who were willing to share their experiences with us. We also thank the research team for their hard work and commitment to ensuring the success of this study. Members of the MD-PREV research group are: Alice Theadom (Chair and Principal Investigator), Kerry Walker, Tamasin Taylor, Miriam Rodrigues, Richard Roxburgh, Gina O’Grady, Annemarei Ranta, Donald Love, Jenny Stewart, Gemma Poke, Graeme Hammond-Tooke, Ronelle Baker, Margaret Williams, Kelly Jones, Braden Te Ao, Priya Parmar, Valery Feigin, and Rita Krishnamurthi. This work was supported by the Health Research Council of New Zealand [grant number 14/399].
Appendices
List of all included conditions and sub-types
| Condition | Sub-types |
|---|---|
| Dystrophinopathies (N = 42) | Duchenne muscular dystrophy (N = 35) |
| Becker muscular dystrophy (N = 7) | |
| Manifesting carriers of Dystrophinopathies (N≤6) | |
| Facioscapulohumeral muscular dystrophy | FSHD1 |
| FSHD2 | |
| Subtype unknown | |
| Emery-Dreifuss muscular dystrophy (N≤6) | EDMD1 (EMD) |
| EDMD2 and 3 (LMNA) | |
| EDMD4 (SYNE1) | |
| EDMD5 (SYNE2) | |
| EDMD6 (FHL1) | |
| Subtype unknown | |
| Limb-girdle muscular dystrophy (N≤6) | Type 1 Autosomal dominant subtypes including; |
| 1A (MYOT), 1B (LMNA), 1C (CAV3), 1E (DNAJB6), 1F (TNPO3), 1G (HNRNPDL) | |
| Type 2 Recessive inheritance including; | |
| 2A (CAPN3), 2B (DYSF), 2C (SGCG), 2D (SGCA), | |
| 2E (SGCB), 2F (SGCD), 2G (TCAP), 2H (TRIM32), | |
| 2I (FKRP), 2J (TTN), 2K (POMT1), 2L (ANO5), | |
| 2M (FKTN), 2N (POMT2), 2O (POMGNT1), 2Q (PLEC1), 2R (DES), 2S (TRAPPC11) | |
| Subtype unknown | |
| Congenital muscular dystrophy (N = 7) | Merosin deficient congenital muscular dystrophy (LAMA2) |
| Selenoproteinopathy (SEPN1) | |
| Laminopathy (LMNA- related CMD) | |
| Alphadystroglycanopathy (POMT1, POMT2, POMGNT1, FKTN, FKRP, and others) | |
| Collagen VI myopathy (COL6A1, COL6A2, COL6A3) | |
| – Ulrich congenital muscular dystrophy | |
| – Bethlem myopathy | |
| Subtype unknown | |
| Distal muscular dystrophy | Laings distal myopathy (MYH7) |
| ANO5-related myopathy | |
| Miyoshi myopathy (DYSF) | |
| Welander (T1A1) | |
| Subtype unknown | |
| Congenital myopathy (N = 11) | Central core myopathies (Central Core Disease and multi-minicore disease) |
| Nemaline myopathy | |
| Centronuclear myopathy (including myotubular myopathy, Congenital fibre type disproportion, Myosin storage myopathy, myotubular myopathy, Other) | |
| Subtype unknown | |
| Myotonic Dystrophy (N≤6) | Myotonic dystrophy (DM1) |
| Myotonic dystrophy (DM2) | |
| Subtype unknown | |
| Other myopathies (N≤6) | Myofibrillar myopathy |
| Oculopharangeal muscular dystrophy | |
| Inclusion body myopathy/GNE myopathy | |
| Subtype unknown | |
| Ion Channel Muscle Disease (N≤6) | Myotonia congenita – Thomsen’s, Becker’s |
| Paramyotonia congenita | |
| Periodic paralysis, Anderson Tawil Syndrome | |
| Subtype Unknown | |
| Pompe disease | |
| Other |
N = numbers of cases in the current study sample. Note: For privacy reasons, N≤6 is reported for those conditions in the current sample with few cases.