
Abstract
Anti-IgLON5-syndrome is known to be an antibody-mediated disease that mainly affects central structures in tegmentum and hippocampus and leads to associated symptoms like sleep-related disturbances, parkinsonism, chorea and limb ataxia. This is the first case of a patient with anti-IgLON5-syndrome and generalized fasciculations and neuromyotonic discharges in electromyography, suggesting that a peripheral involvement may occur in anti-Ig-Lon5-syndromes.
Anti-IgLON5-Syndrome is known to be an antibody-mediated, rapidly progressive disease and mainly comprises sleep-associated symptoms like excessive daytime sleepiness and NREM and REM parasomnia with sleep apnea and stridor, often accompanied by bulbar dysfunction. The syndrome is caused by antibodies against the neuronal cell-adhesion protein IgLON5, that shows a high association with the HLA-DQB1*05 : 01 and HLA-DRB1*10 : 01 alleles. Postmortem studies demonstrate a mainly central disease, inducing a neuronal tauopathy predominantly involving the hippocampal and tegmental structures [1]. The clinical manifestation of the anti-IgLON5-syndrome was recently described by Gaig et al. [2]. In addition to sleep-associated symptoms, chorea, parkinsonism, limb ataxia, ocular and bulbar symptoms and dysautonomia may occur [3, 4]. The exact role of IgLON5 and its physiological way of interacting within the central nervous system still remains unclear [2, 5–7].
We describe the case of a 58-year-old man with sudden onset of generalized fasciculations and in the further course mild to moderate proximal progressive muscle weakness, mild muscle wasting and severe dysphagia. His clinical presentation also included slight left-sided signs for parkinsonism and dyskinesia of his left abdominal muscles. The duration of these intermittent and uncontrolled slow contractions of abdominal muscles were of approx. up to two seconds and sometimes led to a flexion of the upper body. In contrast to this, the patient showed fast twitching fasciculations of upper and lower limbs. Both dyskinesia and fasciculations persisted during sleep. He had no signs for cognitive deterioration, and there was no relevant pre-existing or concurrent disease.
First symptoms the patient recognized were generalized fasciculations without muscle cramps and without weakness. Nine months later, he noticed slight muscle weakness in proximal muscles of upper and lower extremities, that progressed to a mild proximal weakness. In addition, excessive daytime sleepiness was noticed. Concurrent swallowing problems occurred, but progressed rapidly. There were no signs of dysarthria at any time. He developed severe pneumonia 12 months after onset of symptoms and was dependent on prolonged invasive ventilation for two months due to weaning-failure.
Concisely, investigations revealed high titers of IgLON5-antibodys in serum and cerebrospinal fluid and electromyography showed neuromyotonic discharges in muscles of arms, limbs and abdominal muscles (Fig. 1). Conduction velocities of motor and sensory nerves were normal on upper and lower extremities, and repetitive nerve stimulation was normal on trapezoid and orbicularis oculi muscles. In comprehensive electromyography, neuromyotonic discharges with doublets, tri- and multipletts were predominantly seen in proximal muscles of the limbs and abdominal muscles (Fig. 1) in absence of single fasciculation potentials. At slight voluntary muscle contraction, there were no relevant neurogenic or myogenic changes. MRI of brain was normal, particularly without any signs for encephalitis. A DaTSCAN of brain was not performed. In electroencephalography, alpha waves were at 8/s without any signs for epilepsy. Main diagnostic findings in laboratory assessments finally revealed high titers of IgLON5-antibodies in serum (1 : 1.000+++) and cerebrospinal fluid (1 : 100++). Antibodies against VGKC, CASPR2 or LGI1 were absent. There was no evidence for neoplasia in whole-bodyPET scan. Videoflouroscopy revealed severe oropharyngeal dysphagia with poor oral bolus transport, residues in the epiglottic valleculae and in the piriform sinus after swallowing, leading to pre- and postdeglutitive aspiration. There were no clinical signs for affection of the second motor neuron in bulbar muscles (e.g. muscular atrophy or fasciculations). He received nutrition via percutaneous endoscopic gastrostomy.
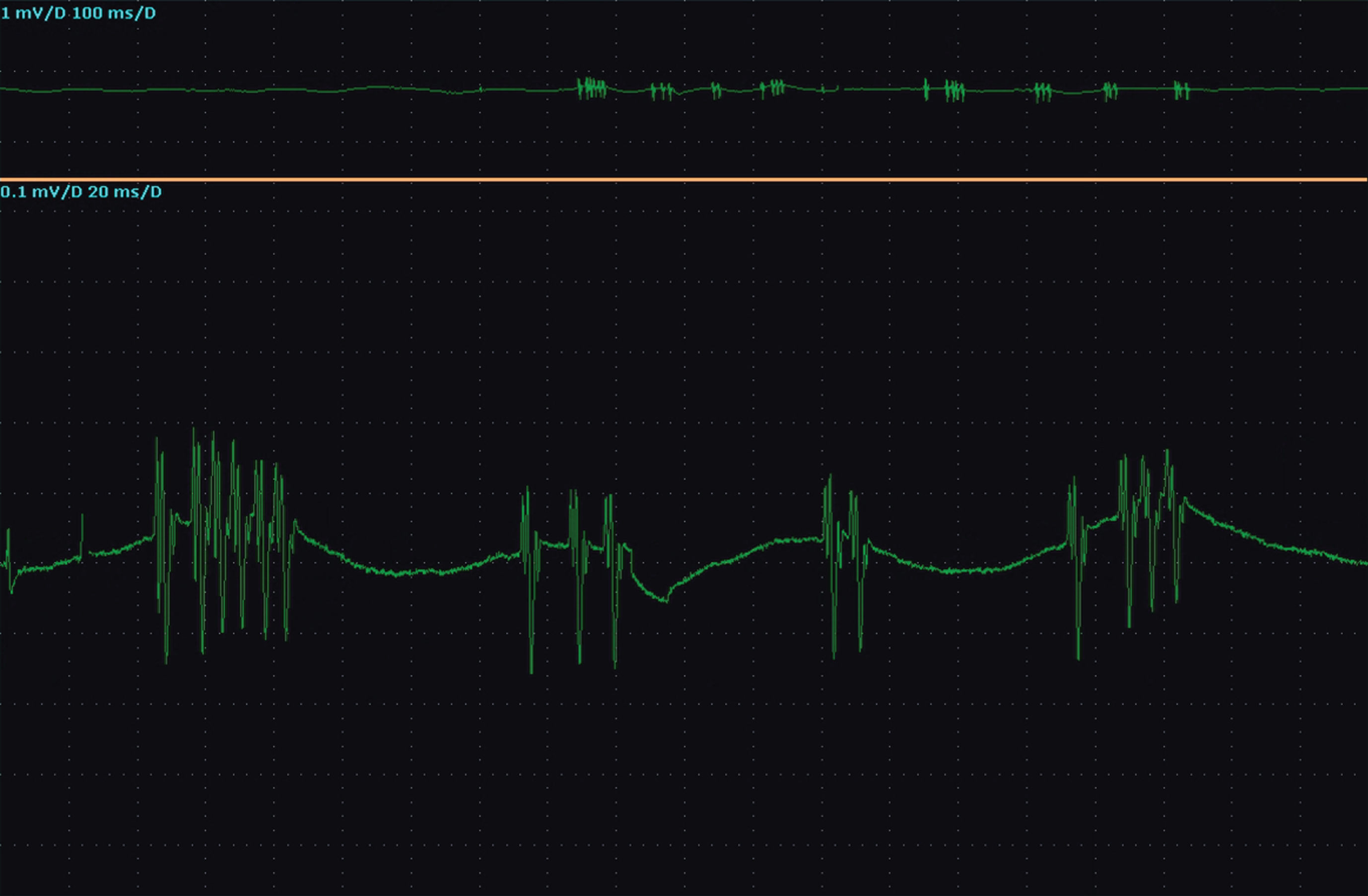
Screenshot of EMG doublets, triplets and multiplets in IOD1 (upper window 1 mV/Div 100 ms/Div, lower window 0.1 mV/Div 20 ms/Div).
When immunosuppressive therapy was initiated with prednisolone 1.500 mg i.v. (three consecutive days 500 mg i.v. per day) and tapered to 15 mg per day p.o., symptoms improved. Clinically, fasciculations and muscle cramps improved slightly, but abdominal dyskinesia did not improve, so carbamazepine was recommended but refused by the patient. Repeated EMG studies did not show any changes of neuromyotonic discharges after therapy. His dysphagia improved slightly but persisted under regularly logopaedics and physiotherapy, so the patient remained mainly on artificial nutrition. For small amounts, oral ingestion was possible. After discharge from the hospital, he had still signs for restrictive respiratory insufficiency but was not NIV-dependent. Due to weak cough, he needed mechanical cough assistance several times a day. In following appointments, his weakness of upper and lower limbs improved moderate by regular physiotherapy. He still complained about mild-to moderate daytime sleepiness. Two years after onset of his first symptoms, the patient had sudden death during sleep. A post-mortem examination has been refused by his wife.
This case illustrates the expanding clinical spectrum of IgLON5-syndrome in this very new, uncommon and not fully understood condition. The exact role of these antibodies and their way of interaction in central and peripheral nervous system remain unclear, but there has been no evidence for a generalized inflammatory or neurodegenerative disorder [2]. In contrast to previously reported cases with sole affection of the central nervous system, this patient had signs for peripheral involvement with neuromyotonic discharges, muscle weakness and wasting in addition to his central syndromes, suggesting an additional affection of the second motor neuron or peripheral nerve. Previously, also Schroeder et al. reported a case of a 77-year old patient where a peripheral involvement of cranial nerves was observed with weakness of facial and bulbar muscles [4]. Fasciculations and peripheral muscle weakness have been reported in two of 20 patients in a retrospective analysis by Honorat et al. [8]. Corresponding to our case, until today there is no published case with signs for central weakness (e.g. spasticity). Therefore, it should be discussed whether this peripheral affection, that caused a hyperexcitability syndrome and peripheral weakness in our case, was primarily or directly induced by IgLON5-antibodies or this IgLON5-syndrome was secondary accompanied by an antibody-mediated Neuromyotonia/Isaacs Syndrome in absence of VGKC- or associated antibodies (CASPR2, LGI1). Usually the response to therapy is poor, and only few cases describe an improvement after immunotherapy or plasmapheresis [5, 6]. In our case, a mild improvement was reached by prednisone regarding dysphagia and muscle weakness including respiratory muscles, but his CNS-related symptoms persisted. The exact cause of his death is unclear, but central sleep apnoea as well as aspiration due to dysphagia must be considered.
With respect to previous published cases, there is evidence that anti-IgLON5 antibodies mainly interact in central nervous system, but a growing number of clinical descriptions also suggest that they may play a minor, but important role on peripheral nerves. Therefore, as long as the full clinical spectrum with its significant variability and exact role of anti-IgLON5 antibodies are not fully described, comprehensive investigation of peripheral and central nervous system should be done accordingly.
AUTHOR CONTRIBUTIONS
Stephan Wenninger did the case interpretation, manuscript drafting and submitted the manuscript.
Co-investigators
n/a
AUTHOR DISCLOSURES
Dr. med. Stephan Wenninger reports no disclosures. Dr. med. Stephan Wenninger has accredited grant agreements by DGM (Deutsche Gesellschaft fuer Muskelkranke e.V.), a part of OPTIMISTIC Trial grant agreement n° 305697, European Community’s Seventh Framework Programme (FP7/2007–2013) and has received speaker honoraria by Recordati Pharma, CSL Behring and RG GmbH.
Footnotes
ACKNOWLEDGMENTS
n/a