
Abstract
Background:
Early-onset myopathies are a heterogeneous group of neuromuscular diseases with broad clinical, genetic and histopathological overlap. The diagnostic approach has considerably changed since high throughput genetic methods (next generation sequencing, NGS) became available.
Objective:
We present diagnostic subgroups in a single neuromuscular referral center and describe an algorithm for the diagnostic work-up.
Methods:
The diagnostic approach of 98 index patients was retrospectively analysed. In 56 cases targeted sequencing of a known gene was performed, in 44 patients NGS was performed using large muscle specific panels, and in 12 individuals whole exome sequencing (WES) was undertaken. One patient was diagnosed via array CGH. Clinical features of all patients are provided.
Results:
The final diagnosis could be found in 63 out of 98 patients (64%) with molecular genetic analysis. In 55% targeted gene sequencing could establish the genetic diagnosis. However, this rate largely depended on the presence of distinct histological or clinical features. NGS (large myopathy-related panels and WES) revealed genetic diagnosis in 58.5% (52% and 67%, respectively). The genes detected by WES in our cohort of patients were all covered by the panels. Based on our findings we propose an algorithm for a practical diagnostic approach.
Prevalences:
MTM1- and LAMA2-patients are the two biggest subgroups, followed by SEPN1-, RYR1- and Collagen VI-related diseases. 31% of genetically confirmed cases represents a group with overlap between “congenital myopathies (CM)” and “congenital muscular dystrophies (CMD)”. In 36% of the patients a specific genetic diagnosis could not be assigned.
Conclusions:
A final diagnosis can be confirmed by high throughput genetic analysis in 58.5% of the cases, which is a higher rate than reported in the literature for muscle biopsy and should in many cases be considered as a first diagnostic tool. NGS cannot replace neuromuscular expertise and a close discussion with the geneticists on NGS is mandatory. Targeted candidate gene sequencing still plays a role in selected cases with highly suspicious clinical or histological features. There is a relevant clinical and genetic overlap between the entities CM and CMD.
Keywords
ABBREVIATIONS
Candidate gene sequencing
Comparative genomic hybridization
Congenital Myopathies
Congenital Muscular Dystrophies
Cardiomyopathy
Exome aggregation consortium
Genome aggregation database
Myotonic Dystrophy Type 1
Next Generation Sequencing
Spinal muscular atrophy
Prader Willi Syndrome
Whole exome sequencing
INTRODUCTION
Early-onset myopathies are a group of inherited neuromuscular diseases. The entire group consists of a wide range of rare diseases with a high clinical and genetic heterogeneity [1–5]. Initially, early-onset myopathies have been differentiated into two large groups based on histological findings:
The classification of congenital myopathies with structural abnormalities is based on typical histopathological findings such as cores, nemaline bodies or central nuclei [4]. There is a broad genetic and histopathological overlap; mutations in a gene can result in different histological phenotypes, and vice versa [4, 9]. The most common histological subtypes are nemaline-myopathy, central core disease, centronuclear myopathy and core myopathy, most frequently due to mutations in the RYR1, MTM1, ACTA1, SEPN1 and NEB genes [10–15]. However, the list of disease causing genes is still growing [16].
The congenital muscular dystrophies (CMD) have been divided into those with pure muscular involvement and those with additional organ involvement, e.g. eye and/or brain abnormalities. Several subtypes are caused by defects in the extracellular matrix, e.g. changes in collagen VI (Col6A1, COL6A2, COL6A3) [17–20], in LAMA2 [5, 21], or defects of nuclear envelope proteins with mutations in genes like EMD or LMNA [2, 22–24]. Furthermore there is a spectrum of disorders in 0-glycosylation (alpha-dystrogylcanopathies) [2].
With further knowledge of the genetic background of these diseases, the limitations of a pure histological classification became evident. A considerable genetic overlap exists between CM with structural abnormalities and CMD. Mutations in the RYR1, SEPN1 and ACTA1 [25] genes are seen in both groups [1, 26]. In some patients with distinct clinical signs and symptoms of a congenital myopathy, histological findings are either minimal (“minimal change myopathy”) or unspecific such as fiber type disproportion. This can be the only morphologic abnormality in conditions like ACTA1, MYH7, RYR1, SEPN1 and TPM-related myopathies and does not allow a clear-cut categorization based on a mere morphological basis [27–29].
The reported frequency of subtypes varies significantly between different centers and regions [3, 31]. Recently, O’Grady et al. evaluated the incidence and the diagnostic outcome of a large cohort of 123 CM/CMD cases [25]. They found muscle biopsy to be inferior to NGS methods in biopsied patients. The study did not include the data from non-biopsied patients, but the authors estimated a probability of detection by genetic methods of about 50% in this group of patients.
In this retrospective study we present the diagnostic approach in a large cohort of patients with early-onset myopathies, seen in a single German referral center. The diagnostic yield of different genetic approaches is discussed and an algorithm for a rational diagnostic work-up under real-world conditions, including financial aspects, is presented.
MATERIALS AND METHODS
Patient cohort
The total cohort of early-onset myopathies included 98 index patients who were regularly seen in the Pediatric Neuromuscular Center at the Dr. v. Hauner Children’s Hospital at Ludwig-Maximilian University of Munich in Germany.
Patients were included according to the following criteria:
All patients showed severe muscle weakness from birth on or beginning within the first year of life. Some children had additional findings like elevated CK-levels, contractures, skeletal deformities, additional brain involvement, skin involvement, respiratory and cardiac involvement. All patients underwent detailed clinical examination by experienced pediatric neurologists, muscle ultrasound screening and neurophysiological examination to confirm myopathy and to exclude a neurogenic disorder. Patients with congenital myotonic dystrophy or congenital myasthenic syndrome were not included in the study. All patients underwent genetic testing, either candidate gene sequencing (direct/targeted sequencing of known genes) and/or NGS (whole exome sequencing or large myopathy-related panels). In 57 patients muscle biopsy was performed prior to genetic testing; in one patient muscle biopsy followed NGS.
Molecular genetic analysis
After written informed consent, genomic DNA was extracted from EDTA-blood samples, according to standard procedures for candidate gene sequencing via Sanger sequencing or multiplex ligation-dependent probe amplification (MLPA).
For the myopathy-related next generation sequencing panels, genomic DNA was extracted with the FlexiGene DNA Kit following the manufacturer’s protocol (http://www.qiagen.com/). Target enrichment was performed with the HaloPlex Target Enrichment System (Protocol Version D, Agilent Technologies, Santa Clara, CA, USA) with an input of 50 ng DNA. After library pooling, next-generation sequencing was performed on the Illumina MiSeq System (Illumina, San Diego, USA) with 150 base pair (bp) paired-end reads and 7pM library input. An average coverage of at least 300 x of the target region including coding exons and 25 bp of flanking intronic sequence was aimed. The sequence analysis was evaluated by using the software SeqPilot of JSI (http://www.jsi-medisys.de/) including the module SeqNext. Generated fastQ-files were aligned to the human reference genome hg19/GRCh37.
Between 2013 and 2017, the panel was constantly updated and 5 different versions of the panel have been used (Fig. 1).
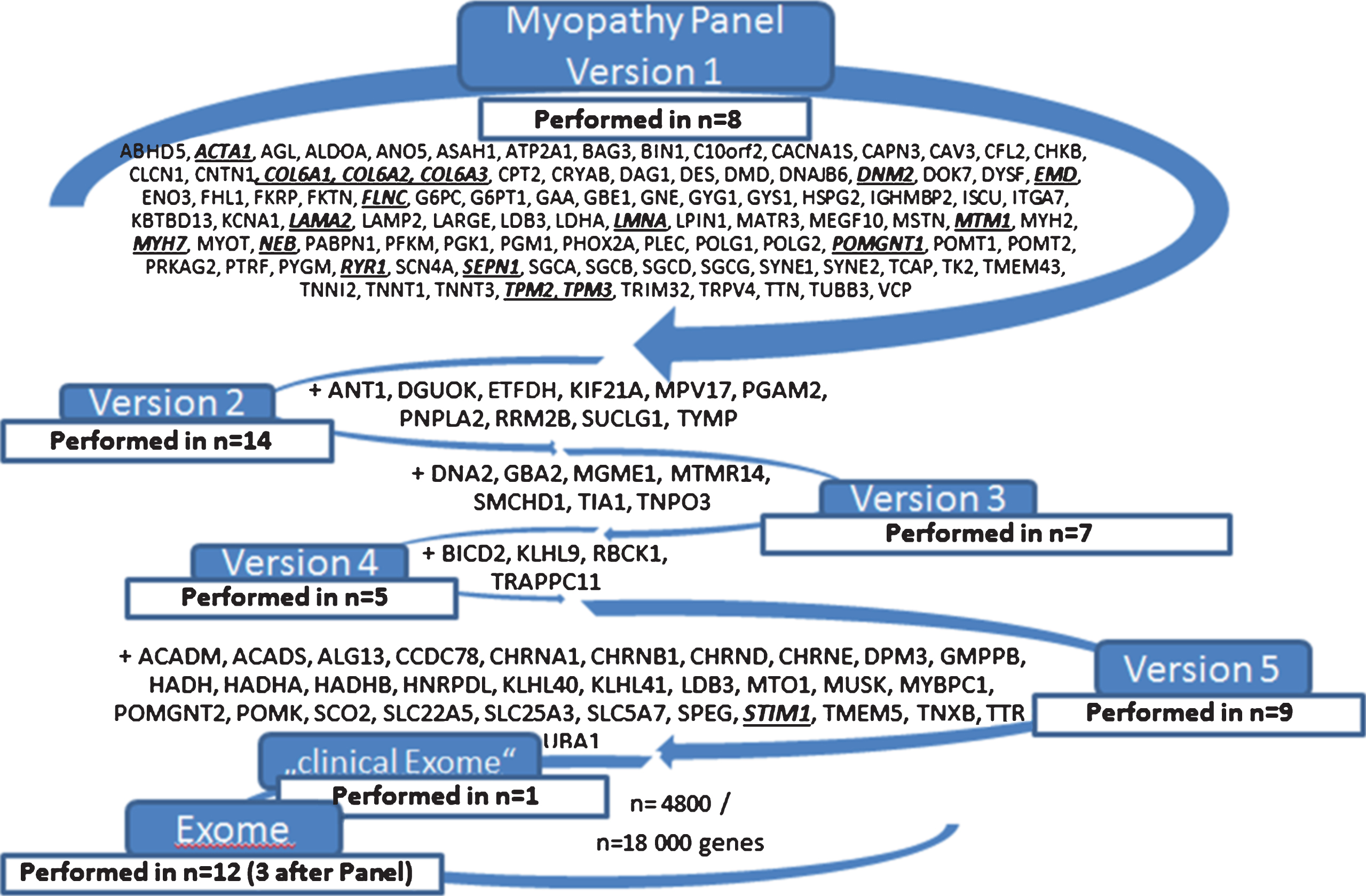
The different next generation methods, revealing genetic diagnosis in 63 index patients in 17 different genes (italic, bold and underlined).
Exome sequencing and variant filtering was done as described previously (28), using a SureSelect Human All Exon 50Mb V5 or SureSelect Human All Exon 60Mb V6 Kit (Agilent, Santa Clara, CA, USA) for enrichment of coding DNA sequences and a HiSeq2500 or HiSeq4000 system (Illumina, San Diego, CA, USA) for sequencing. Reads were aligned to the human reference assembly (hg19) using BWA (version 0.5.8) and single-nucleotide variants (SNVs) and small insertions and deletions were identified with SAMtools (version 0.1.7). Insertions and deletions were detected with pindel and ExomeDepth. The average coverage was 131-fold and 97% of the targeted regions were covered at least 20-fold.
The analysis was carried out considering the following modes of inheritance: For an autosomal dominant inheritance a search for genes carrying heterozygous potentially pathogenic rare DNA variants (minor allele frequency, MAF <0.2%) was performed. Accurate clinical phenotyping was done in close communication with the referring clinician. A search in available literature and databases (OMIM, Pubmed, ClinVar, HGMD) was additionally used to identify genes suitable for the according phenotype. For an autosomal recessive inheritance rare variants with a MAF of less than 1.0% are considered (homozygous or compound heterozygous). Frequencies are evaluated according to our in-house database and with the ExAC and gnomAD databases, containing respectively more than 60,000 and 135,000 samples. All modes of inheritance are considered for sporadic cases given the possibility of de novo mutations as the underlying genetic explanation for disease. Evaluation of pathogenicity was done according to the guidelines by the American College of Medical Genetics after segregation analyses in families when feasible.
The following panels have been used (Fig. 1): NGS myopathy panel Version 1 in 8 patients, Version 2 in 14 patients, Version 3 in 7 patients, Version 4 in 5 patients, and Version 5 in 9 patients. A panel comprising 4800 known disease-causing genes was performed in 1 patient and WES was done in 12 patients (including 3 patients with previous myopathy-related panel).
Levels of significance were calculated using the Fisher Test for 2×2 Contingency Table.
The study was approved by the ethics committee of the University of Munich (UE 141-14).
RESULTS
Molecular genetic analysis
The whole cohort of 98 patients underwent genetic testing. Figure 2 provides a flow diagram to visualize the step-by-step diagnostic workup for all patients. Biopsy findings and clear phenotypic data that guided the targeted testing are mentioned in detail. 58 of all patients had a muscle biopsy, 57 before and one after genetic testing. Molecular genetic methods (targeted candidate gene sequencing and NGS methods together) led to a confirmed genetic diagnosis in 63/98 patients (64%). Array-CGH disclosed a large homozygous deletion comprising the COL6A1 gene in one patient (patient 13, Supplemental Table 1). Array CGH in this patient was initiated by another hospital due to unclear motor delay. High throughput genetic analysis (next generation sequencing panels and whole exome sequencing) was done in 53 patients. Twelve of them had targeted candidate sequencing before, four of them array CGH, and in three of them WES followed a panel, but remained without diagnosis. The methods confirmed genetic diagnosis in 31 of the 53 patients (58.5%). In detail, WES was leading to the diagnosis in 67% (8/12) and myopathy-related panels in 52% of the cases (23/44).
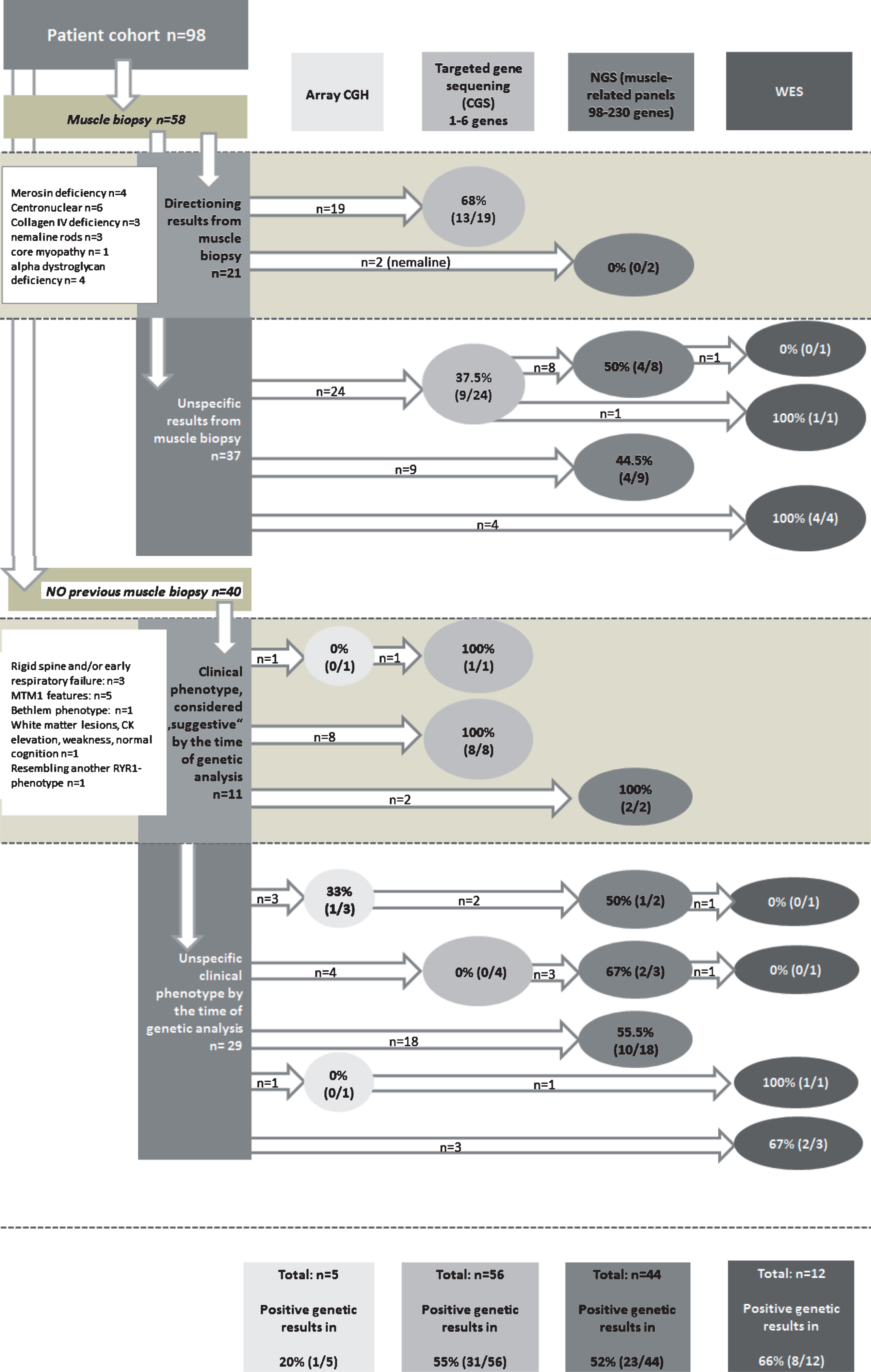
Flow diagram to visualize the step-wise diagnostic workup and the number of positive genetic results (absolute number/%) for all patients. Biopsy findings and clear phenotypic data that guided the targeted testing are mentioned in detail and in their frequency in the white boxes.
Targeted candidate gene sequencing was performed in 56 patients and established diagnosis in 31 of the 56 (55%). Sanger Sequencing of up to 6 single genes was only successful in 9 of 24 patients with non-specific biopsy findings (37.5%), but in 13 of 19 patients with an informative muscle biopsy (68%), such as a negative immunostaining of a protein or a distinct structural abnormality (p < 0.01). In patients without a previous muscle biopsy but with suggestive clinical phenotype, the result rate of candidate gene sequencing was 100% (8/8 patients). In these patients distinct clinical features directed the search for specific genes. Most often a typical symptom complex was severe floppy infant syndrome, respiratory failure combined with ophtalmoplegia and/or cryptorchidism in four MTM1-patients. Early respiratory failure and/or rigid spine were the symptoms in three SEPN1-patients. One patient showed white matter lesions in a CT scan (performed due to an accidental fall) which was directive for a LAMA2-mutation. One patient with a “Bethlem”-phenotype was assigned via targeted gene sequencing. COL6A3 mutation was confirmed after previous negative Sanger sequencing of COL6A1 and COL6A2 genes.
All detailed results from muscle biopsy, genetic testing prior to the finally diagnostic test and failed testing results are presented in Supplemental Table 2. Supplemental Table 1 provides detailed clinical data such as CK levels, distribution of weakness, course of the disease, orthopedic features, respiratory and cardiac involvement and result from muscle ultrasound-screening. With these data in mind and also considering cost efficacy, we created an algorithm for diagnostic approach (Fig. 3). It considers specific clinical findings and, if available findings from biopsy.
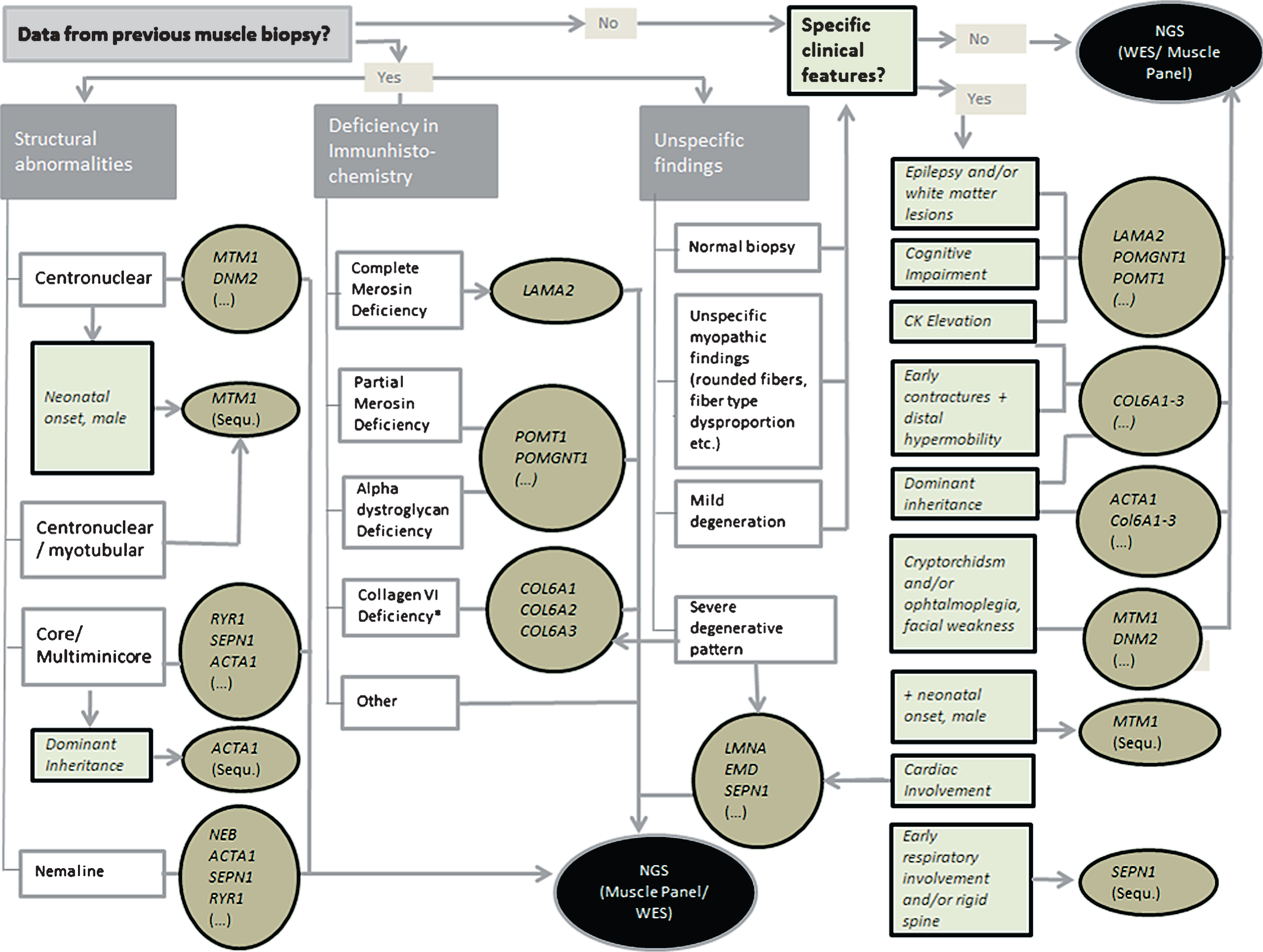
Algorithm for the diagnostic approach. Clinical features and the most common genetic differential diagnosis are mentioned especially for the discussion with the analysing geneticist. NGS = Next generation sequencing.
Disease frequencies
Within our cohort of 98 index patients, 63 patients (64%) have a genetically confirmed diagnosis with mutations in the genes listed below in descending order:
LAMA2 (n = 10), MTM1 (n = 10), SEPN1 (n = 7), RYR1 (n = 7), Col6A1 (n = 5), Col6A2 (n = 4), Col6A3 (n = 3), LMNA (n = 3), NEB (n = 1), ACTA1 (n = 3), DNM2 (n = 2), TPM2 (n = 1), TPM3 (n = 1), EMD (n = 1), MYH7 (n = 1), FLNC (n = 1), POMT1 (n = 1), POMGNT1 (n = 1), STIM1 (n = 1) (Fig. 4). One ACTA1-patient and his brother had been described before ([15], patient II:6 and III:2). One index patient from the family with autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation together with his father, has recently been published [35].
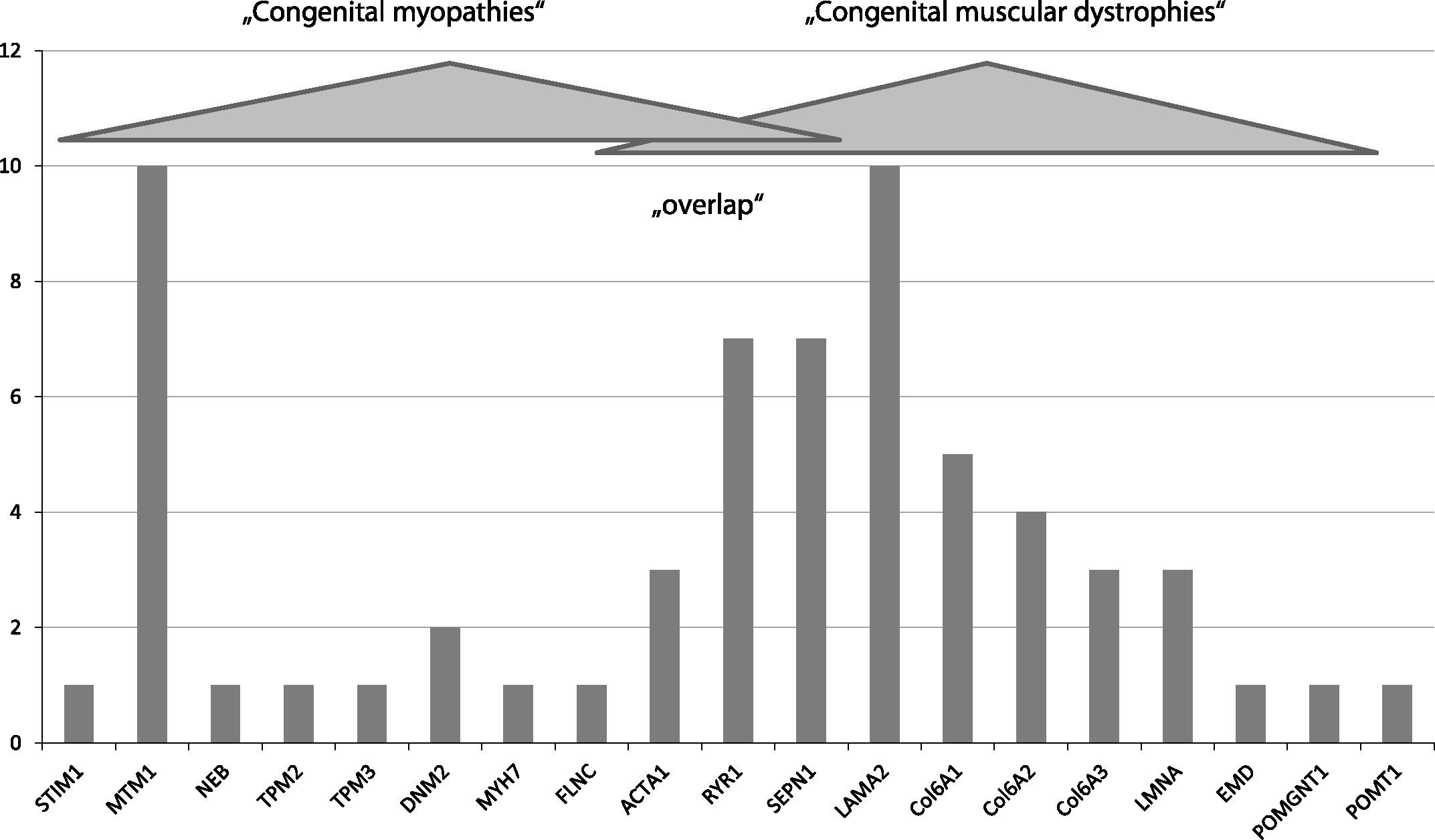
Relative prevalence in our cohort.
Three patients showed an alpha-dystroglycan deficiency indicating a disorder of O-glycosylation without genetic confirmation despite targeted candidate gene sequencing. 36 patients (patients 61–94, 20, 44, Supplemental Table 1), remained without a confirmed genetic diagnosis. 13 of them had targeted candidate gene sequencing only. Most of these patients were lost to follow-up at our clinic before the appearance of NGS.
The clinical (and histological if applicable) features of the 22 patients without genetic diagnosis despite NGS methods are listed in Supplemental Tables 1 and 2 (patients 61, 63, 65, 66, 67, 68, 69, 76, 77, 82–94). This group is very heterogeneous and comprises patients with all features of early-onset myopathies. Included are 9 patients with a benign course of mild proximal or axial weakness. There is one patient with severe scoliosis, one with a “classic” rigid spine syndrome and one with impressive hypermobility and hypertrophic scars. Two patients have predominant respiratory problems. Two have dilated cardiomyopathy as the major clinical finding. One had a most severe neonatal weakness showing rods in the muscle biopsy. One showed proximal weakness and neck contractures combined with epilepsy and intellectual disability. Two had severe proximal weakness, and one a distal myopathy, hyperlaxity of joints and a narrow face.
DISCUSSION
We retrospectively studied the diagnostic yield of genetic testing in a cohort of 98 index patients with early-onset myopathies. We present the incidence of various subgroups in a single German academic center (Fig. 4), the diagnostic approach (Fig. 2, Supplemental Table 2), and all relevant clinical data including CK levels, distribution of weakness, contractures, skeletal deformities, additional brain involvement, skin involvement, respiratory and cardiac involvement and result from muscle ultrasound-screening (Supplemental Table 1). We present an algorithm for genetic testing (Fig. 3). All patients were followed in our referral center for pediatric neuromuscular disorders indicating the relative prevalence of different disease subgroups in southern Germany.
In most neuromuscular centers the search for a genetic diagnosis has become the standard. Due to the advances in molecular genetics, muscle biopsy is more and more replaced by genetic testing as first line diagnostic tool in patients with clinical signs of CM or CMD.
Role of NGS, targeted candidate sequencing and clinical expertise
The diagnostic algorithm for early-onset myopathies has considerably changed during the past few years. With the appearance of new genetic techniques such as high-throughput analysis (next generation sequencing such as myopathy-related panels, whole exome sequencing or genomic sequencing [2, 25] [36]) less invasive techniques have partially replaced muscle biopsy as a first diagnostic step. We found that in 64% of our patients genetic testing (targeted candidate gene sequencing and next generation sequencing methods together) confirmed the clinical diagnosis of a congenital myopathy or a congenital muscular dystrophy. When we correlate our findings to the data presented recently by O’Grady this rate is significantly higher than the one reported from muscle biopsy (p < 0.1) ([25, 36].
The detection rate of myopathy-related panels and WES was 58.5%, a percentage which is in the same range as reported by other groups [2, 38]. Targeted candidate gene sequencing was leading to the diagnosis in 55% (31 of 56). This rate is similar to the one in NGS. Looking at the group of biopsied patients, this rate holds only true for those with a highly specific muscle biopsy, especially with absent merosin (laminin-alpha2) or a myotubular myopathy. It is much lower in patients with non-specific histological findings. In the cohort of non-biopsied patients, the same was true for those with an informative clinical phenotype like rigid spine syndrome or severe neonatal onset of weakness with dysmorphic features. Three of our SEPN1 patients and four of our MTM1 patients were directly diagnosed by sequencing possible candidate genes according to clinical suspicion. This underlines the role of careful clinical assessment despite the tremendous progress in molecular genetic testing, and mirrors the importance of neuromuscular expertise, as recentlyreported [39].
In close cooperation with the geneticist it has to be discussed even in cases with a strong clinical hypothesis which technical approach is the most promising and the least expensive. This is especially true for large genes like RYR1 or LAMA2 where NGS is much cheaper than Sanger sequencing.
In our experience the successful use of high throughput genetic analysis requires a close cooperation between pediatric neurologist, pathologist and the molecular geneticist. We have established a regular neuromuscular-genetic board to discuss the clinical phenotype, to build a hypothesis, to check if potentially causative genes are sufficiently covered by the NGS method and to crosscheck whether molecular genetic findings could explain the clinical phenotype.
In our cohort, either large myopathy-related panels including all the muscle genes which were known at the time of examination or WES was used. All the pathogenic genes found by WES were also covered by the myopathy-related panels. For this reason WES as second line diagnostic procedure after the use of large myopathy-related panels cannot be recommended in patients with yet undetermined congenital myopathies. Since coverage is still a challenge in NGS [25, 40] smaller panels including the most frequent causative genes (Fig. 3) might be a useful first step. If negative, WES or a large muscle-specific panel should follow.
The disease-causing mutations in all 63 clarified patients were found in 17 genes, only.
However, there is still a considerable part of the patients who lack a genetic diagnosis. Since newly recognised genes are responsible for the disease in at least 5% of patients [25] and many new genes will be identified in the future the use of WES which allows a reanalysis of data might be preferable. In the future whole genome sequencing (WGS) which is at the moment only available on a scientific basis might help to elucidate further cases.
Array-CGH can be considered before performing WGS to exclude a large deletion containing muscle genes. In our cohort, only five patients had array CGH, which was diagnostic in one Col6A1-patient.
Interpretation of new variants of unclear significance
One of the biggest challenges of high throughput genetic techniques, is the interpretation of changes of yet unknown significance in some patients. In these cases a muscle biopsy or a second look at previously done histological examinations can help to clarify the situation like in patient No. 39 (Supplemental Table 1), where a variant of unclear significance (c.13927G>A (p.Ala4643Thr)) was identified in the RYR1 gene, a rare SNV (single nucleotide variant) (reported 6 times in ExAC), which was also detected in the unaffected father. A second look at the biopsy revealed discreet multi-minicores, confirming the diagnosis of multi-minicore disease. Given this histopathological diagnosis, there are two potential explanations: The first would be autosomal dominant inheritance with incomlete penetrance which we consider rather unlikely or autosomal recessive inheritance in compound heterozygosity with a second, so far unidentified mutation in our patient.
Nevertheless histological finding can be misleading. In two patients with partial merosin deficiency (patients No. 26 and 96, Supplemental Table 1) in muscle biopsy an O-glycosylation defect was proven. One patient had POMGNT1-related CMD, the other had POMT1-related CMD. There are only few case reports of a secondary merosin deficiency, among them already one patient with a mutation in the POMT1 gene [32].
One MTM1-patient with symptoms and signs compatible with a myotubular myopathy (weakness of proximal and facial muscles, ophthalmoplegia and cryptorchidism) (No. 53, Supplemental Table 1; mutation c.528G>A, identified in WES) showed a splice site mutation. Muscle biopsy was atypical, showing only fiber type disproportion and mitochondrial pathology. No central nuclei were found in the biopsy. His clinical course is milder than usual.
Rare clinical phenotypes
Patients with early-onset myopathy and cardiomyopathy were rare in our cohort. Six patients had an associated cardiomyopathy, five underwent heart transplantation. Two of these had LMNA mutations, which is a frequent association. These are in line with the literature regarding the cardiac phenotype [33], but unusual for the skeletal weakness, which was improving over time. One patient with dilative CMP had a MYH7 mutation. Another patient with dilative CMP showed a FLNC mutation; a de-novo variant, so far not described and of unclear relevance. So far FLNC mutations have only been described with restrictive CMP [34]. Two patients with dilated CMP could not be clarified by the multigene myopathy panel version 2. However, some newer CMD-associated genes like TNNT2, MYBPC3 or LDB3 were not included in that panelversion.
Prevalence
The relative frequency of subtypes in our cohort (Fig. 4) differs from reports of other centers. Especially the number of RYR1-patients was lower compared to the study of Maggi et al. [3]. Similar differences between regions and centers have been reported in the literature [30, 31].
25% of our genetically determined patients with early onset myopathies belong to the group formerly defined as “congenital myopathy with structural abnormalities”. Patients with mutations in the MTM1 gene (myotubular myopathy) are the biggest subgroup.
44% of the patients with genetic diagnosis were diagnosed as “congenital muscular dystrophy”, with mutations in the LAMA2-gene (merosin-negative CMD) representing the largest subgroup. This is consistent with other previously reported findings. A Danish study recently calculated a LAMA2-mutation frequency of 28% in the CMD population [41].
31% of our patients with genetic diagnosis belong to a group which is in the literature no longer clearly defined as CM or CMD. This is especially true for patients with mutations in SEPN1 and RYR1. According to the literature we found an overlap between CM with structural abnormalities and CMD in these genetic defects [1, 26] (Fig. 1).
Conclusions
Combining deep clinical phenotyping, muscle imaging and molecular genetic analysis has become the major pathway for the diagnosis of early onset myopathies.
A stepwise approach to a child with clinical signs of a congenital myopathy/muscle dystrophy as outlined in Fig. 3 has to take into account the clinical phenotype, the histological findings if available and economic aspects such as costs of various genetic approaches. Targeted candidate gene sequencing still plays a role in selected cases with highly suspicious clinical or histological features. If very large genes are suspected based on clinical or histological findings, like collagen VI-subtypes or RYR1, high throughput techniques are preferable. Neuromuscular expertise and close cooperation between geneticist and clinician is indispensable. In the future, the role of muscle biopsy will change from a first step diagnostic tool to a confirmation tool for genetic changes with yet unknown significance, or for patients without genetic diagnosis but clear clinical suspicion. There is a relevant genetic and clinical overlap between the former entities CM and CMD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
AUTHOR’S CONTRIBUTION
Vill and Müller-Felber wrote the manuscript; Vill, Müller-Felber, Schroeder and Blaschek cared for the patients; Vill, Blaschek, Walter, Schoser, Schlotter-Weigel, Gerstl, Tacke, Borggraefe and Müller-Felber performed the neuromuscular diagnostic work-up; Schoser and Müller-Felber reviewed the biopsies, Gläser and Kuhn performed the NGS myopathy panels, Haack, Wagner, Kovacs and Alhaddad performed the Whole Exome Sequencing, Müller checked for English style and language, all authors reviewed the manuscript.
Footnotes
Supplemental Tables 1 and 2 are available in the electronic version of this article: ![]() .
.