
Abstract
Background:
Spinal muscular atrophy (SMA) is a neurodegenerative autosomal recessive disorder characterized by the loss of α-motor neurons. A variety of molecular pathways are being investigated to elevate SMN protein expression in SMA models and in the clinic. One of these approaches involves stabilizing the SMNΔ7 protein by inducing translational read-through. Previous studies have demonstrated that functionality and stability are partially restored to the otherwise unstable SMNΔ7 by the addition of non-specific C-terminal peptide sequences, or by inducing a similar molecular event through the use of read-through inducing compounds such as aminoglycosides.
Objective:
The objective was to determine the efficacy of the macrolide Azithromycin (AZM), an FDA approved read-through-inducing compound, in the well-established severe mouse model of SMA.
Methods:
Initially, dosing regimen following ICV administrations of AZM at different post-natal days and concentrations was determined by their impact on SMN levels in disease-relevant tissues. Selected dose was then tested for phenotypic parameters changes as compared to the appropriate controls and in conjugation to another therapy.
Results:
AZM increases SMN protein in disease relevant tissues, however, this did not translate into similar improvements in the SMA phenotype in a severe mouse model of SMA. Co-administration of AZM and a previously developed antisense oligonucleotide that increases SMN2 splicing, resulted in an improvement in the SMA phenotype beyond either AZM or ASO alone, including a highly significant extension in survival with improvement in body weight and movement.
Conclusions:
It is important to explore various approaches for SMA therapeutics, hence compounds that specifically induce SMNΔ7 read-through, without having prohibitive toxicity, may provide an alternative platform for a combinatorial treatment. Here we established that AZM activity at a low dose can increase SMN protein in disease-relevant animal model and can impact disease severity.
Keywords
INTRODUCTION
Spinal Muscular Atrophy (SMA) is an autosomal recessive disorder characterized by increasing muscle weakness and loss of lower motor neurons. SMA is the second most common autosomal recessive neurodegenerative disorder with an incidence of 1 in 10,000 worldwide [1–3] and a carrier frequency of ∼1 in 40 [4]. Disease progression leads to atrophy of voluntary muscle groups, paralysis and premature death for many patients. Despite a range of disease phenotypes (SMA Type I – Werdnig-Hoffmann disease OMIM 253300; SMA Type II – Dubowitz Disease OMIM 253550, SMA Type III – Kugelberg-Welander disease OMIM 253400, SMA Type IV – Adult Onset Form OMIM 271150), SMA is caused by loss or mutations in a single gene called Survival Motor Neuron 1 (SMN1). The patient population is homogeneous due to loss or mutation in the telomeric copy of the SMN1 gene, but all SMA cases rely on the nearly identical copy gene, SMN2, which produces low levels of the full-length, functional SMN protein [5]. SMN is ubiquitously expressed and present diffusely within the cytoplasm and in punctate structures in the nucleus. SMN is a critical factor in a variety of RNA pathways, including its essential role in the formation of spliceosomal small nuclear ribonucleoproteins (snRNPs). Even though the SMN2 gene is 99% identical in nucleotide sequence, approximately 90% of SMN2-derived transcripts are alternatively spliced and encode a truncated protein lacking the crucial coding exon 7. This aberrant splicing event is the result of a silent, non-polymorphic C to T nucleotide transition 6 nucleotides within exon 7 [6, 7].
Translation termination occurs when one of the three stop codons, UAA, UGA and UAG, enters the ribosomal A-site, where it is recognized and dissociates by the release factors eRF1 and eRF3 [8]. It has long been appreciated that pharmacological agents such as aminoglycoside antibiotics can reduce the fidelity of translation termination by a mechanism that alters the ribosomal proofreading process. Additionally, the degree of translational infidelity is significantly influenced by the codon and surrounding sequences [9]. Read-through therapy has been envisioned for a variety of diseases, often genetic disorders that arise from premature termination codons, including Duchenne and Becker muscular dystrophy (DMD & BMD), cystic fibrosis (CF), and Hurler syndrome [9–18]. Despite the fact that pharmacologically-induced read-through is an enticing molecular strategy, several aminoglycosides have shown off-target toxicity in clinical studies [19]. Read-through efficiency in vivo and toxicity have been concerns for aminoglycosides; however, novel compounds such as Ataluren have advanced to late stage clinical trials for DMD and CF [20–23]. Similarly, several macrolide antibiotics with known safety profiles were shown to induce read-through of disease-causing stop codons in the ATM gene in ataxia-telangiectasia (A-T), MeCP2 gene that result in Rett syndrome (RTT), the SMN2 gene in SMA, and mutations in the adenomatous polyposis coli (APC) gene that lead to familial adenomatous polyposis (FAP) [24].
The SMN2 gene is an excellent target for therapeutic intervention since it retains the coding capacity for the wildtype SMN protein. A variety of SMA therapeutic strategies have targeted SMN2, including: promoter stimulation; modulating exon 7 splicing with antisense oligonucleotides or small molecules; stabilizing the SMN transcript; and stabilizing the SMN protein [25, 26]. The notion of stabilizing the SMNΔ7 protein is based on the observation that SMNΔ7 protein functionality can be improved by increasing the length of the C-terminal peptide or by genetically altering the amino acid composition of a degradation sequence unique to the SMNΔ7 protein [27–30]. Consistent with this notion, read-through inducing compounds such as aminoglycosides have been shown to increase SMN protein levels and decrease disease severity in SMA mouse models [27, 29–33], yet their discovered toxicity encourages the search for safer read-through agents.
In this report, a FDA-approved drug with known read-through activity was investigated as a potential therapeutic either as a stand-alone therapy or in combination with a potent splice-switching ASO in a severe SMA mouse model (SMNΔ7). Azithromycin monohydrate (AZM) belongs to the macrolide antibiotic family of the polyketide class and consists of a large macrolide ring to which one or more deoxy sugars are attached. The lactone ring can be either 14, 15 or 16 membered [34]. The mechanism of action for these compounds is through inhibition of bacterial protein biosynthesis by preventing peptidyltransferase from adding the growing tRNA attached peptide to the next amino acid, thus hindering ribosomal translation [35, 36]. We tested the effect of AZM as a read-through mediator to induce SMN expression in the severe SMNΔ7 (Smn–/–;SMN2+/+;SMNΔ7+/+) model [37]. These results demonstrate that as a stand-alone therapy at low doses, the severe SMA phenotype was very slightly improved following AZM treatment, while the splice-switching ASO was better but only mildly active. However, when used in combination, a significant improvement in the SMA phenotype was observed.
MATERIALS AND METHODS
Azithromycin monohydrate preparation
Azithromycin Monohydrate (Batch No.: 711168643510513) was manufactured by Pliva Croatia Ltd. (Pliva Hrvatska d.o.o.; Prilaz Baruna Filipovića 25; 10000 Zagreb, Croatia, EU) and supplied by BioBlast Pharma Ltd. (37 Dereh Menachem Begin St.; Tel Aviv 6522042, Israel). The monohydrate is a white powder of 767.5 MW and 98.7% purity. The Azithromycin was kept in a 4°C refrigerator, protected from light, under desiccant. The formulated concentration (stock solution) was 10 mg/ml and stored in 4°C refrigeration, protected from light following preparation, until the time of use. The expiration date for the stock solution was set at one week following preparation with the freshly dosing solution used on the same day.
Azithromycin monohydrate dose preparation
Preparation of Azithromycin Monohydrate stock solution of 10 mg/ml was conducted according to the following procedure: Azithromycin Monohydrate – 100 mg; 1.5 mmol/mL Sodium Phosphate monobasic (NaH2POP4, Fisher Scientific, Cat. No. BP330-500) – 400μL; Sodium chloride (NaCl, Fisher Scientific, Cat. No. BP358-1) – 0.057 g; ddH2O (Milli-Q® Ultrapure Water System, MilliporeSigma, Merck, KGaA, Darmstadt, Germany) up to 10 mL. The stock solution of the Azithromycin Monohydrate is isotonic and at pH range of 6.4–6.8 (typically around 6.5). Following preparation, stock solution was filtered through 0.2μm sterilized filter. AZM formulations were prepared by further dilutions with normal Saline (0.9% NaCl) that served also as the vehicle control.
Animal procedures and AZM delivery in the severe SMA mouse model
All animals were housed and treated in accordance with either the Animal Care and Use Committee guidelines of the University of Missouri, Columbia, MO, USA; or the National Institutes of Health’s Guide for the Care and Use of Animals, approved by Institutional Animal Care and Use Committee (IACUC) of The Jackson Laboratory, Bar Harbor, ME. Animals were fed low-fat stock diets (Harlan Teklad 8640). The colony was maintained as heterozygote breeding pairs under specific pathogen free conditions. SMNΔ7 (SMNΔ7+/+;SMN2+/+;Smn–/–; JAX® Stock #005025: FVB.Cg-Grm7Tg (SMN2) 89Ahmb Smn1tm1Msd Tg(SMN2*delta7)4299Ahmb/J) [37] were genotyped on the day of birth (P0) using standard PCR protocol (JAX® Mice Resources) on tail tissue material. The following primer sets were used: for the mouse Smn gene, mSmn-WT forward (5’-tctgtgttcgtgcgtggtgacttt-3’) and mSmn-WT reverse (5’-cccaccacctaagaaagcctcaat-3’) and for the Smn knockout SMN1-KO forward (5’-ccaacttaatcgccttgcagcaca-3’) and SMN1-KO reverse (5’-aagcgagtggcaacatggaaatcg-3’). Intracerebroventricular (ICV) injections were performed on P1, as previously described [38–41]. Briefly, mice were immobilized via cryoanesthesia and injected using μL calibrated sterilized glass micropipettes. The injection site was approximately 0.25 mm lateral to the sagittal suture and 0.50–0.75 mm rostral to the neonatal coronary suture. The needles were inserted perpendicular to the skull surface using a fiber-optic light (Boyce Scientific Inc.) to aid in illuminating pertinent anatomical structures. Needles were removed after 5 seconds of discontinuation of plunger movement to prevent backflow. Treated animals were placed in a warmed container for recovery (5–10 minutes) until movement was restored. A single injection of 7μl of AZM was delivered via ICV as described above for all mice. Body weight and life expectancy were monitored and recorded daily. Time-to-right (TTR) reflex tests were conducted as previously described [42]. Each pup was placed onto its back and the time it takes to right itself on the ground was recorded. The test was terminated at 30 seconds and if an animal had not turned by this time, it was recorded as ‘Failure’. Righting reflex measurement were recorded daily starting at P7 since unaffected animals start to turn over at this time. At each occasion, three attempts were recorded separately. The Hind Limbs Suspension (HLS, Hind Legs Spread or Tube Test) test and score are an overall evaluation of the hind-limb spread (in cm) during the first 10–15 seconds of hanging onto the lip of the tube, after which the observer does not change the score even if the animal shows a lower or higher score at a later time during the test. If the animal falls within 5 seconds, the HLS score is evaluated based on this time period. The effect of fatigue on the HLS score is normally captured in subsequent trials [43]. HLS was measured every other day between P8-P20.
ASO preparation and administration
ASO delivery was performed by intracerebroventricular (ICV) injection using methods described previously [41]. The antisense oligo (MOE1v11) was synthesized with morpholino modifications at every nucleotide position (GeneTools L.L.C.; Philomath, OR 97370 USA). A previously established suboptimal baseline of the MOE1v11 ASO resulting in a modest extension of survival was administered at P1 via 5μL injection consisting of 2 nanomoles of the oligo [44].
Western blots
For the SMNΔ7 mouse Western blots, indicated tissues were collected at P6 and immediately frozen in liquid nitrogen. Tissue samples were placed at – 80°C until ready for analysis. Roughly 100 mg of tissue was homogenized in Jurkat Lysis Buffer (JLB) (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 20 mM NaH2(PO4), 25 mM NaF, 2 mM EDTA, 10% glycerol, 1% Triton X-100, and protease inhibitors (Roche, Indianapolis, IN, USA)). Equal amounts of protein were separated on 12% SDS-PAGE gels. SMN immunoblots were performed using a mouse SMN specific monoclonal antibody (BD Biosciences, San Jose, CA, USA) diluted 1 : 300 in TBST (Tris-buffered Saline Tween20 (10 mM Tris-HCl, pH7.5, 150 mM NaCl, 0.2% Tween20)) in 1.5% dry milk. Then blots were visualized by chemiluminescence on a Fujifilm imager LAS-3000 (FujiFilmUSA, Hanover Park, IL, USA) and the corresponding software. To verify equal loading, membranes were re-probed with rabbit Anti-IP90 primary antibody (IP90, anti-Calnexin, Catalog #PA5-34665, Invitrogen™ Thermo Fisher Scientific, Carlsbad, CA, USA) and anti-rabbit HRP secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA), diluted 1 : 2,000 and 1 : 10,000, respectively. Western blots were performed in triplicates or more and representative blots are shown. The signal intensity was measured for each band on an immunoblot, normalized to the loading control, and the fold increase was determined in relation to the appropriate control. Data were quantified utilizing FUJI FILM MultiGauge v. 2.0.
ELISA determination of SMN protein expression
Brain, spinal cord and muscle tissues were extracted from treated animals at the time of necropsy. Tissues were analyzed for total SMN by enzyme-linked immunosorbent assay (ELISA) (SMN ELISA Kit, Enzo Life Sciences; Farmingdale, NY, USA) according to the manufacturer’s protocol. Values were normalized per milligram of total protein. Due to the low number of tissues and the high variability among them, preventing even outliers’ omission, statistical analyses using several statistical software and methods were performed (Supplementary Table 4).
Statistical analysis
Statistical analysis and calculations were performed utilizing several software and analytical methods: SAS software suite v9.3 (SAS Institute, SAS Campus Drive, Cary, North Carolina 27513, USA). Kaplan-Meier survival curves for the study groups were compared with a Log-rank test (GraphPad Prism v5.00; GraphPad Software, Inc., 7825 Fay Avenue, Suite 230, La Jolla, CA 92037 USA, or SAS software). The tube test and weight change from baseline were analyzed with repeated measures ANOVA (SAS PROC MIXED; SAS/STAT(R) 9.2, SAS Institute, SAS Campus Drive, Cary, North Carolina 27513, USA), where each was modeled as a function of group (Untreated, Healthy, Vehicle Treated and AZM Treated), time and the group by time interaction term. LSMeans (Least Squares Means) differences between the groups per time point were estimated from the model interaction terms and are presented with respective levels of significance and 95% confidence intervals. The level of significance presented assesses whether the LSMean difference is significantly different from 0 (zero). In addition, LSMeans for each group per time point were estimated from the model interaction terms and are presented with 95% confidence intervals in graphs. A p-value of ≤0.05 was considered statistically significant. For SMN expression (ELISA), statistical analyses were performed by GraphPad Prism as above (2-way ANOVA with Tukey’s multiple comparisons), by Student’s t-test (of each AZM dose vs. vehicle treated) using Excel 2013, version 15.0.4753.1003 and by IBM SPSS Statistics software. Body weight analysis of combined therapy was done using repeated measures mixed model analysis, comparing untreated to either AZM- or ASO-treated or comparing AZM- to AZM/ASO-treated.
RESULTS
Analysis of SMN protein levels in the SMNΔ7 mouse model of SMA following Azithromycin monohydrate delivery
Azithromycin monohydrate (AZM) is a FDA approved compound that has the potential to serve as a stand-alone or as a combinatorial therapy, particularly since the mode-of-action for AZM is through a read-through event (Fig. 1A) which could provide an additive benefit for compounds that increase SMN transcription or splicing. The goal of the initial phase of this study was to determine whether ICV injections of AZM increase SMN expression levels in vivo. SMNΔ7 animals were randomly enrolled into study groups following genotyping at day of birth (P0). The study was conducted using two doses, a low (5.0 mg/kg) and a high (25.0 mg/kg) AZM dose at the first or second stage, respectively. Compound administration was performed at postnatal days 1–5 (P1-P5) according to the study design tables (Supplementary Tables 1 and 2). Protein homogenates from brain and spinal cord tissues were prepared and separated by SDS–PAGE. SMN was detected by Western blotting using monoclonal antibodies for SMN and a loading control, calnexin. As expected, SMN levels were significantly lower in tissues from SMA mice compared to healthy animals. The results demonstrated increased SMN expression levels in all treated animals in the case of single and multiple administrations (Fig. 1B-D). In the low dose AZM treated cohorts, SMN levels appeared low in brain tissue, with a detectable increase observed in all treated animals (Vehicle and AZM) (Fig. 1B), a phenomenon that was previously observed [45, 46]. SMN levels in spinal cord tissue from low dose injected animals increased in all treated animals compared to the untreated SMNΔ7. An increase in dosing did not improve SMN expression since higher expression was observed in the single (P1), double (P1&P2) and the cohort that received five daily consecutive (P1-P5) administrations. This study supported the notion that SMN protein levels in SMNΔ7 mouse model are increased at the spinal cord due to treatment with AZM, hence we next examined its impact upon the well-characterized SMA phenotype using a single administration of the 5.0 mg/kg dose.
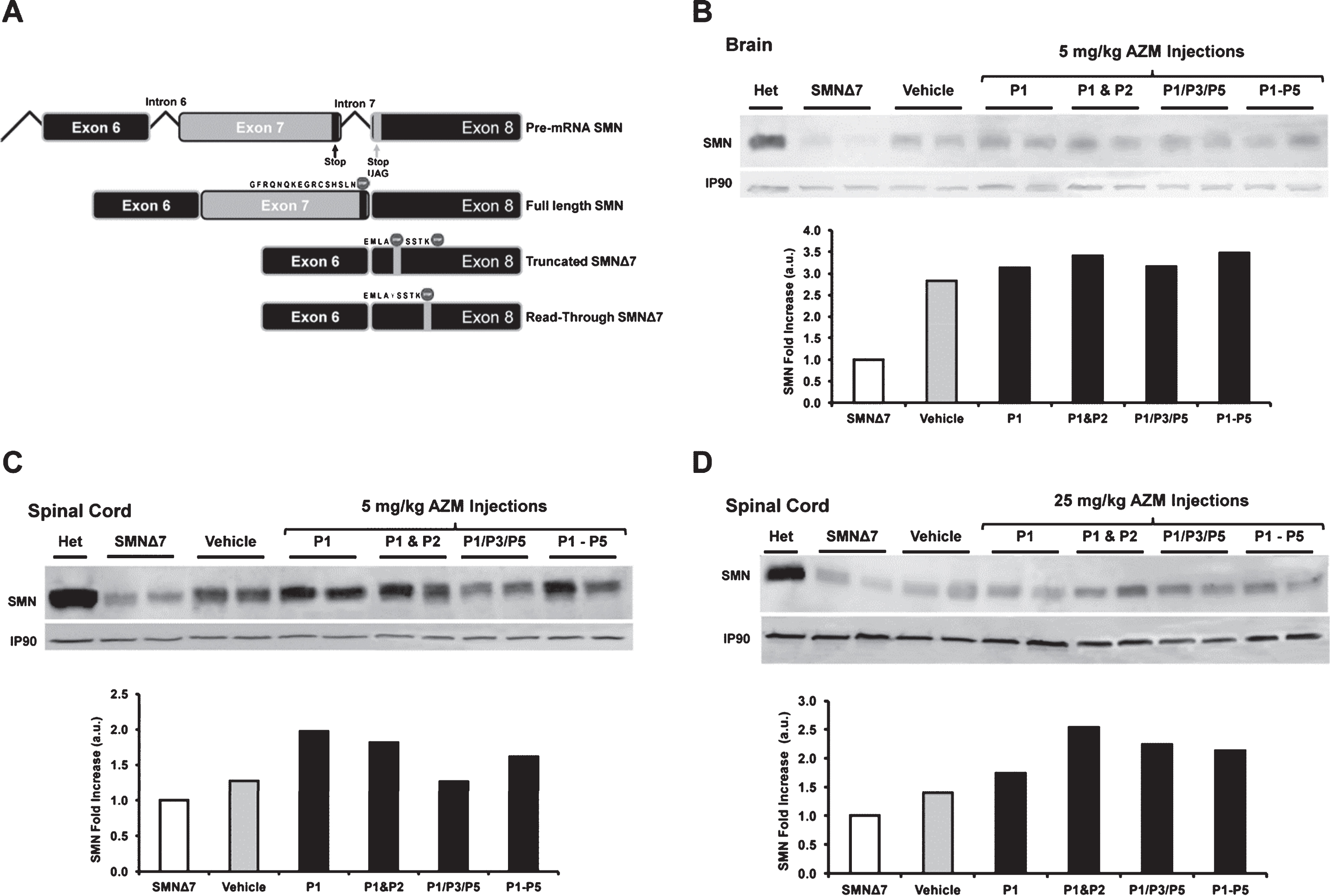
(A) Schematic of compound induced SMN read-through event. The top diagram indicates the 3’ portion of pre-mRNA of SMN and the splicing process. The second line represents the full-length SMN product showing the translational stop codon at the 3’ end of exon7. The third line shows the truncated SMNΔ7 isoform of SMN mRNA with the translational stop codon 16 nucleotides into exon8. The fourth line depicts the SMN read-through product triggered by compound treatment. The mechanism of read-through agents involves altering the fidelity of the ribosome such that a tyrosine is incorporated into the first translational stop codon due to the coding of the wobble position, resulting in a read-through event until the second stop located further downstream. Relevant peptide sequences are shown on the schematics. Western blot images showing increased SMN levels in brain (B), spinal cord (C) tissues after 5 mg/kg AZM treatment; and in spinal cord tissues after 25 mg/kg dose (D). AZM treatment regimens at post-natal days (P) are represented as a single injection (P1), two injections (P1&P2), three q.a.d. injections (P1/P3/P5), and five consecutive injections (P1-P5); vehicle (Saline); untreated (SMA); and healthy (Het). Anti-calnexin (IP-90) was used as internal control. Each lane represents one animal. Bar graphs represent Western blots quantifications. Fold increase in SMN protein induction was compared to the untreated control group and plotted.
Low doses of AZM did not extent survival and did not significantly improve the phenotype of severe SMA mice
To examine the potential for any phenotypic changes following delivery of a low dose of AZM to SMNΔ7 SMA mice, survival, weight gain, righting reflex and hind legs gap measurements were collected following a single ICV injection at P1. At the 5.0 mg/kg dose, the average life span in AZM treated animals was 13.7, an extension beyond either vehicle treated or untreated SMA mice (11.2; 12.3, respectively) (Fig. 2A). Additionally, the longest lived animal was 19 days in the AZM cohort compared to 16 or 17 days; however, the improvement in life span was not statistically significant based upon a Mantel-Cox survival analysis. Similarly, analysis of body weight in AZM treated mice demonstrated that they were not statistically heavier compared to untreated and vehicle treated controls (Fig. 2B). The righting response is an additional established measure of the SMA phenotype designed to measure gross motor function [42]. In this assay, mice were placed on their backs and the time it takes to stand on four legs was recorded daily starting from P7 onwards. Inability to perform the test within 30 seconds was marked as a failed attempt. As anticipated, unaffected (“Heterozygous”) animals were able to right within 1–3 seconds, whereas only 2 animals from the untreated or vehicle treated SMA animals were able to right (Fig. 2C, D). It is notable that on P10 two AZM-treated mice were able to right themselves within 15 seconds. Nevertheless, the results from the righting experiments showed no significant improvement overall (Fig. 2C, D).
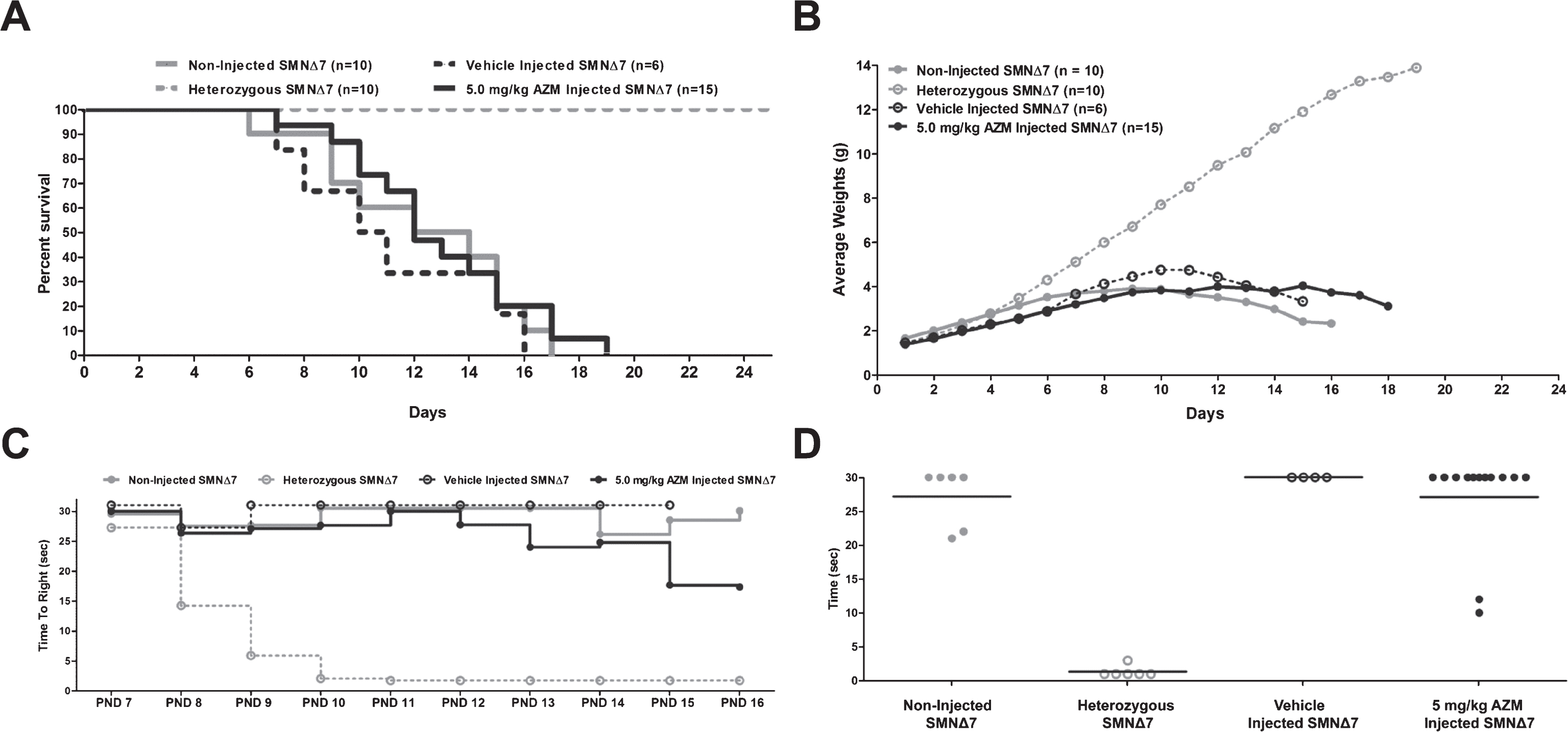
Phenotypic changes in the severe SMNΔ7 mouse models of SMA after 5 mg/kg AZM treatment. (A) Kaplan-Meier survival curves were obtained from the various treatment groups as indicated. Log-rank (Mantel-Cox) statistics were applied for comparisons of AZM vs. untreated animals, and AZM vs. vehicle group. Severe SMNΔ7 SMA mice showed no significant improvement in survival and longevity after injections with AZM. (B) AZM treatment does not result in a significant weight gain. (C) Assessment of overall fitness and muscle function of treated SMNΔ7 animals. Graph representing raw data of the average time to right from P7 to P16. Animals injected with AZM (5 mg/kg dose) did not display improvement in motor function through their life span. Time to right was measured daily from P7 until mortality. (D) Scatter plot of time to right ability of mice injected with AZM highlights the performance of individual mice (values are shown for P10).
Azithromycin (AZM) mechanism of action is based on its ability to interfere to the activity of the protein translation machinery, namely the entrance of the releasing factors eRF1 and eRF3 to the ribosome. As such, contrary to the general notion, dose increase may be harmful to the cell, while lower doses might be more efficacious. We presumed that the results at the 5 mg/kg dose, in which SMN protein was elevated yet only modest changes in the SMA phenotype were observed, may trigger the interaction with the translational machinery and, therefore, lowering the dose may improve the response. Thus, the following experiments were performed with two lower doses of AZM: 1.0 and 0.5 mg/kg. First, we determined whether SMN levels were changed in disease-relevant tissues using more sensitive monitoring system, the commercially available SMN ELISA. To test the effect of AZM on SMN protein levels, disease-relevant tissues such as brain, spinal cord and muscle were harvested from severe SMNΔ7 animals that were treated on P4 with either AZM or vehicle. Brain, spinal cord and muscle tissues were harvested from treated animals at the time of the earlier of 10% body weight loss from their peak body weight, followed by ELISA analysis for SMN content. Animals treated with AZM expressed higher levels of SMN protein in a dose-related manner, as compared to vehicle treated animals in each of the three tissues examined (Fig. 3 & Supplementary Table 3).
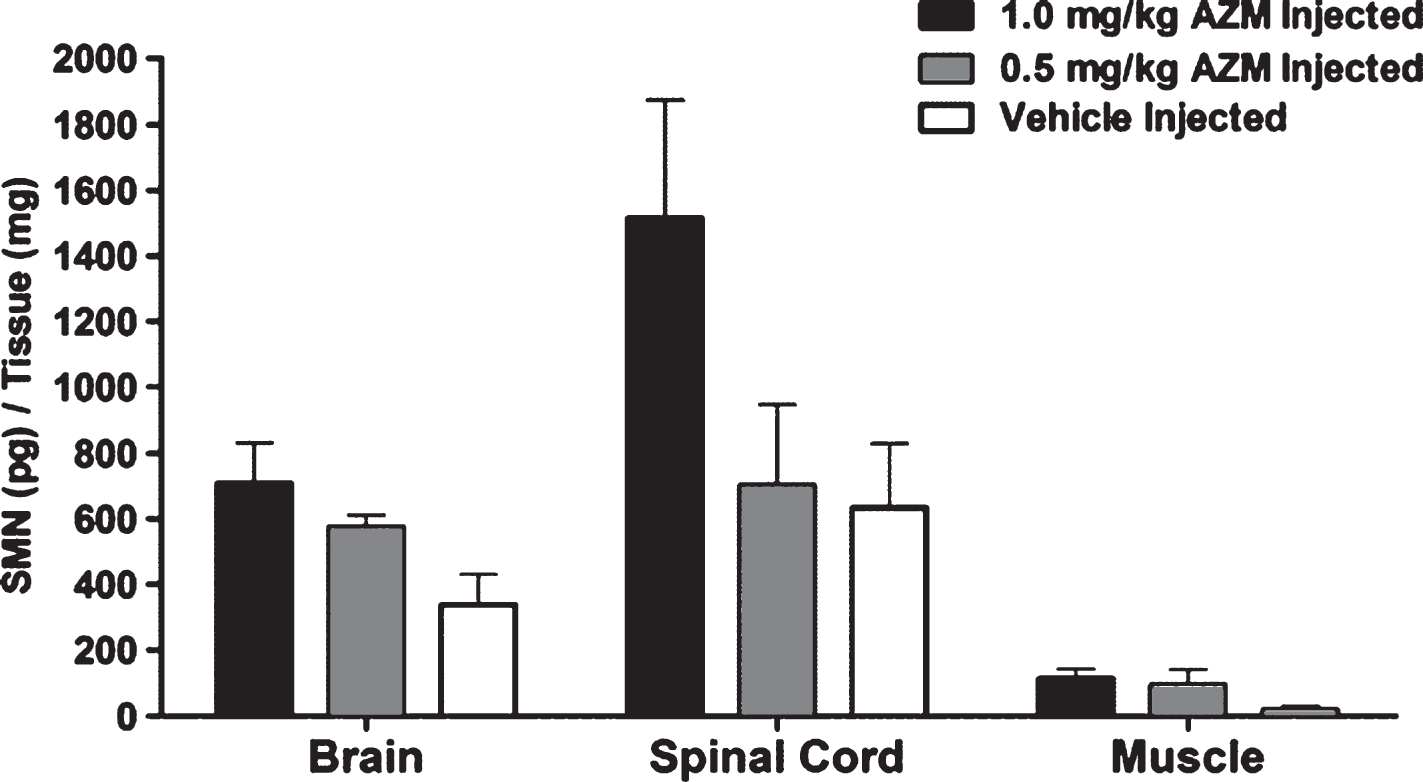
SMN levels in brain, spinal cord and muscle of SMNΔ7 mice. SMN level (pg/mg protein) in brain, spinal cord and muscle obtained from AZM (0.5 mg/kg, n = 4 or 1.0 mg/kg; n = 5) or vehicle treated (Normal Saline, n = 4) animals. Results of pg SMN corrected for mg tissue protein are presented as mean±SEM. Statistical analyses are presented in Supplementary Table 3.
Having established AZM activity at the SMN protein level, we next examined whether a low dose administration of AZM altered the SMA phenotype. SMNΔ7 cohorts received a single ICV-administration of 0.5 mg/kg dose AZM at P1 and were monitored daily for survival (Fig. 4A), body weight (Fig. 4B), righting reflex (Fig. 4C, D) and hind limb spread (Fig. 4E, F). By lowering the dose of AZM to 0.5 mg/kg, we could observe improvements in four disease parameters in the treated animals, including extended survival, greater weight gain, higher percentage of animals that were able to right themselves and an increase in the distance between hind limbs. Specifically, the median survival time in the AZM treatment group was 15.0 days compared to either vehicle treated or untreated SMA mice which lived 10.0 or 13.0 days, respectively (Fig. 4A). Despite the observation that the longest lived AZM treated animal lived to 20 days, the differences were not statistically significant (p = 0.1171, Mantel-Cox Test). Daily body weight measurements for each cohort demonstrated increased weight in the AZM-treated group compared to untreated or vehicle treated SMA pups (Fig. 4B). Statistical analysis for each specific day indicated the increases were statistically significant for the AZM treated group on several days between P8 and P15 (Supplemental Table 3) based upon least squares means (LSMeans) comparisons of SMA untreated or vehicle treated groups vs. AZM treated group per time point. The righting response was examined daily starting on P7 and at each occasion, three attempts were recorded separately as represented by the data on P13 (Fig. 4C). On P13, vehicle treated mice failed to turn and only 3 successful attempts were recorded for the untreated SMA pups, compared to the AZM treated group in which 5 SMA pups were able to right themselves on the first attempt, ranging in speed from 5 to 17 seconds (Fig. 4C). The percent of all treated animals able to right themselves also improved in the AZM treatment group compared to SMA untreated and vehicle treated animals. This difference was statistically significant (p = 0.018) for the AZM compared to the vehicle-treated group (Fig. 4D). An additional characteristic of the SMA phenotype is the decreased splay in the hind limbs when measured in a “tube-test.” To examine whether the AZM improves hind limb splay, each cohort was analyzed in the tube test [43], which measures the distance between the hind limbs (Fig. 4E, F). Distance between hind legs was measured q.a.d. between P8-P20 for the AZM treated (0.5 mg/kg dose, n = 15), vehicle treated (saline, n = 15), untreated (Non-Injected SMNΔ7, n = 10) and unaffected (Heterozygous, n = 10) animals. The average hind limb splay of AZM treated animals was improved compared to vehicle-treated animals (p = 0.036) (Fig. 4E). Pair-wise comparisons of the LS Mean differences between the treatment groups per time point were calculated highlighting the distribution of the hind leg spread for each of day measured (Fig. 4F). The unaffected group gained strength with age as expected, and increased hind leg spread from P8 to P20; however, the remaining SMA cohorts groups showed a continual decrease from P8 in hind limb spread. On P12 there was a statistically (p = 0.0360) greater (0.43 cm) hind leg spread in the AZM versus the vehicle treated group (Supplementary Figure 1). Collectively, these results demonstrated that as a mono-therapeutic agent, low doses of AZM displayed slight impact upon several disease phenotype parameters, but not on the utmost one, namely, lifespan.
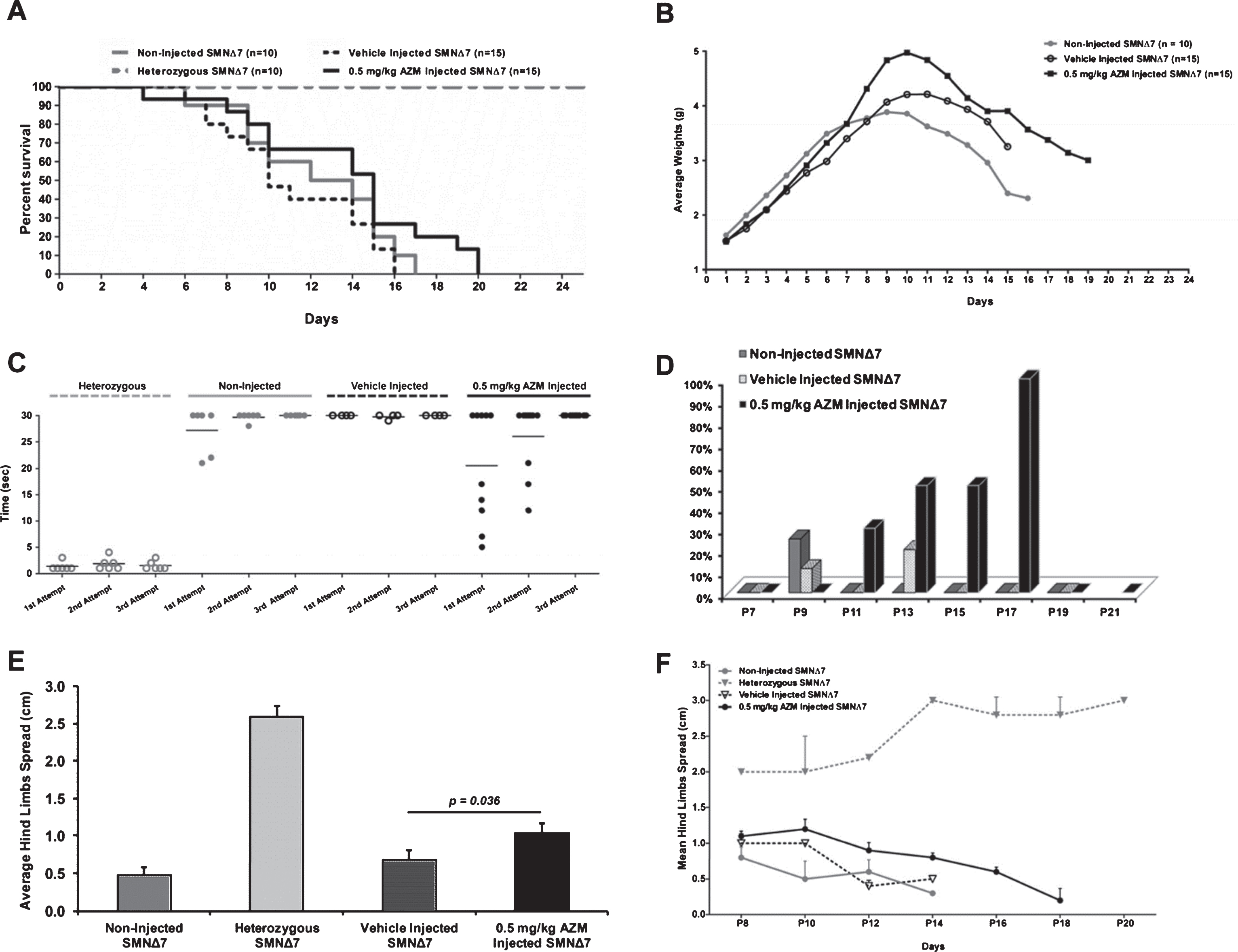
Phenotypic changes in the severe SMNΔ7 mouse models of SMA after 0.5 mg/kg AZM treatment. (A) Life span is presented in a Kaplan-Meier survival plot for AZM treated 0.5 mg/kg dose (n = 15), vehicle treated (Saline, n = 15), untreated (n = 10) and healthy (n = 10) animals. At this lower dose, slightly extended survival was observed in AZM treated animals in comparison to controls, but the positive trend was statistically not significant (p = 0.2394 for AZM vs. untreated; p = 0.0552 for AZM vs. vehicle; p = 0.3888 for vehicle vs. untreated). (B) Average body weight of untreated, vehicle- and AZM-treated SMNΔ7 mice. Animals from the treated group significantly gained more weight than the untreated group during study days P8 – P15 (Supplementary Table 3). Motor function and muscle strength show significant improvement in AZM treated SMNΔ7 mice. (C) Average time to right on P13 for all experimental groups. Distribution of the time to right is presented for each group in three attempts. In the first attempt almost 50% of mice in the AZM treated group were able to right themselves ∼ 16 seconds, whereas about 30% up to 22 seconds in the untreated and less than 10% by 30 seconds in the vehicle treated. In the second and third attempts the time to right gets slower and less frequent in the three groups. (D) Bar graph representing the percent of the tested animals able to right themselves compared to untreated mice where the righting reflex is delayed. Higher percentage of the AZM treated animals was able to right themselves in comparison to untreated and vehicle treated animals. This difference was statistically significant (p = 0.018) for the AZM treated vs. vehicle treated group. (E) Hind legs spread of SMNΔ7 mice ICV treated with 0.5 mg/kg AZM. The distance between the hind limbs of AZM treated animals was significantly larger than the gap of vehicle treated animals (p = 0.036), indicating a slower neurodegenerative process in these animals. (F) Hind leg spread LSMeans with 95% confidence limits per group and corresponding time point. The healthy group trends toward an increase in hind leg spread from P8 to P20, with other groups showing a decrease. Only on P12 there is a statistically significant (p = 0.0360) higher hind leg spread in the AZM treated versus the vehicle treated group.
Combinatorial treatment: AZM and a potent splice-switching antisense oligonucleotide
Modulating SMN2 splicing is an important therapeutic strategy in SMA as evidenced by the recent approval of SPINRAZATM (Biogen Idec/Ionis Pharmaceuticals). It is likely that with the approval of a splice-switching ASO, adjunctive therapeutics could be used to enhance efficacy, particularly those compounds that function through different mechanisms other than exon 7 splicing, i.e. AZM. To determine whether co-administration of a potent SMN2-targeting ASO and AZM further decreased disease severity, a single ICV injection of a previously developed antisense oligonucleotide was delivered to SMA pups at P1. This phosphorodiamidate morpholino oligomer (PMO) molecule targets the E1 regulatory region in SMN2 and is called E1v1.11. For these experiments, a previously identified sub-optimal dose of 2 nanomoles of the E1v1.11 ASO was used to modestly but significantly improve the SMA phenotype in SMNΔ7 animals as a baseline [44], thereby allowing additional benefits from AZM treatment to be observed. Similarly, AZM was administered at 5-fold lower concentration (0.1 mg/kg). Despite its low levels, AZM was able to increase body weight by 0.29gr above untreated mice weight [CI95% (0.103795–0.478690) p = 0.003], while ASO was more effective, increasing body weight by 0.36gr [CI95% (0.142865–0.586702) p = 0.001]. However, addition of AZM to ASO added 0.47gr to mice body weight in comparison to ASO alone at extremely significant level [CI95% (0.323241–0.620821) p < 0.001]. The increase in body weight was mainly observed from P12 to P18 (Fig. 5A). Correspondingly, the righting reflex was enhanced in the AZM/ASO group as determined by the successful attempts/total attempts ratio from P16-P23 (Fig. 5B). The most profound impact of the combinatorial treatment was observed in the life span of the SMA mice (Fig. 5C). Statistical analysis of survival showed that there is a significant difference between all groups [log-rank = 29.1; p < 0.001) with median survival of 11, 14, 16 and 20d for the untreated, AZM-, ASO- and AZM/ASO-treated mice, respectively. Furthermore, while first death was recorded at the first half of the study for no or mono-treatments (P6, P8 and P10 for untreated, AZM- and ASO-treated mice, respectively), the first death in the AZM/ASO treatment group was recorded much later, at P19, demonstrating that the co-treatment also prevented early deaths. Collectively, these results demonstrate that AZM treatment has no effect when administered as single agent; however, when administered in combined therapy with a potent splice-switching ASO, it significantly extends survival and improves disease phenotype.
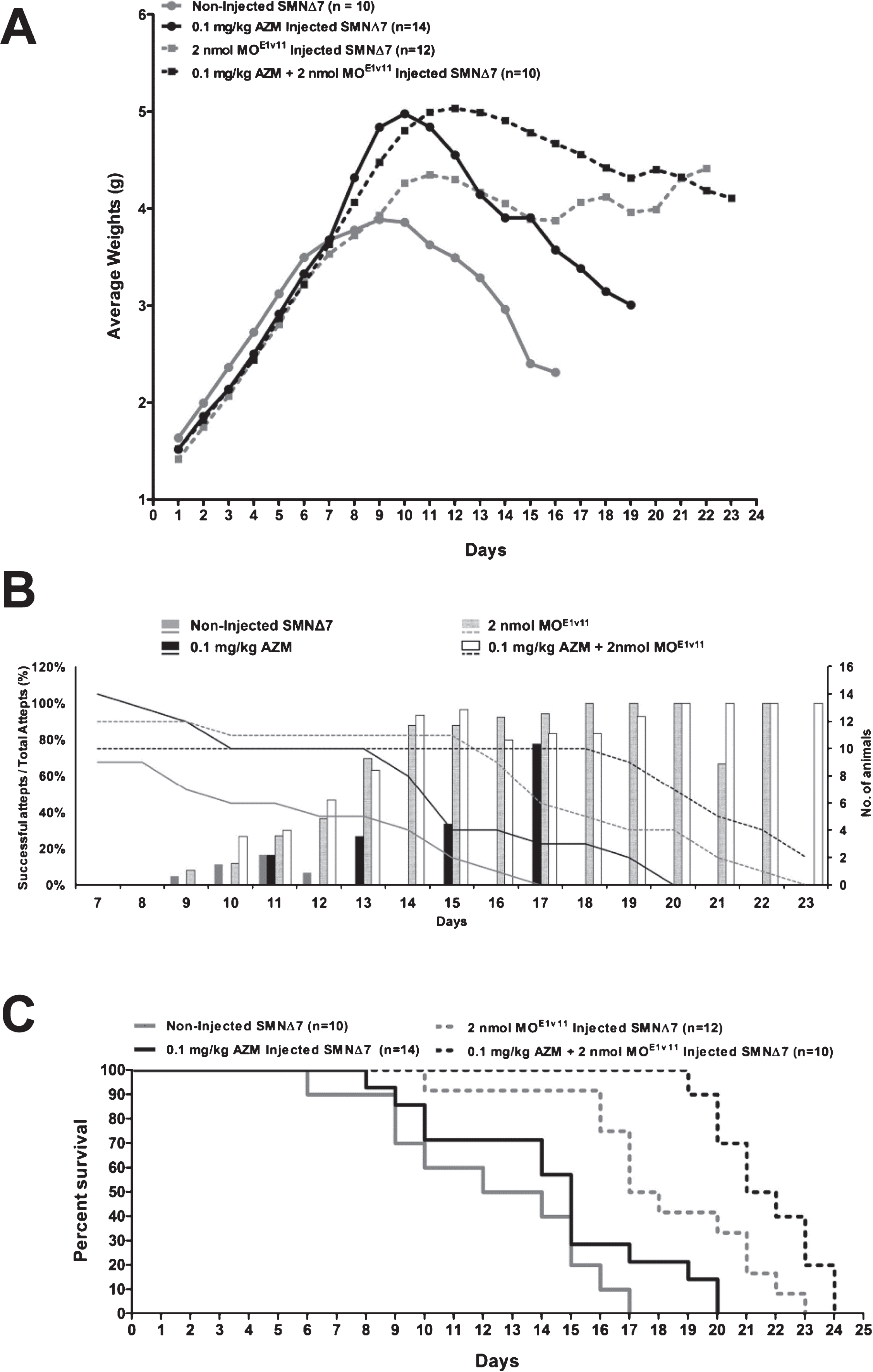
Assessment of a combinatorial treatment with AZM and a potent SMN2-targeting ASO improves disease severity. (A) Weight gain of severe SMNΔ7 mice treated with 0.1 mg/kg AZM dose and a suboptimal ASO (2 nanomoles of MOE1v11) injected once at P1 alone or in combination. Body weight results are presented as a daily mean±SEM. Average weight gain was significantly higher in AZM and ASO alone (p < 0.05), and even better in the combinatorial treated cohorts (p < 0.0001) “p” values were calculated using either Student’s t-distribution two tail measurements or repeated measures mixed model analysis. (B) Percent successful attempts (3 consecutive per individual animal) measurements for the righting reflex showing a significant improvement in muscle function in animals treated with the 0.1 mg/kg AZM after ASO injection compared to controls. (C) Kaplan-Meier survival plot showing the life span for AZM treated 0.1 mg/kg dose (n = 14), MOE1v11 ASO treated (n = 12), untreated (n = 10) and MOE1v11 ASO + AZM treated (n = 10) animals. The co-treated animals showed significant extension in life span in comparison to controls where p < 0.0001 for co-treated vs. untreated; p = 0.016 for co-treated vs. MOE1v11 ASO alone; p < 0.001 for co-treated vs. AZM alone (Log-rank Mantel-Cox test).
DISCUSSION
Due to the presence of SMN2 and the ability to induce fully functional SMN expression from this important back-up gene, a variety of therapeutic approaches have been examined in SMA contexts, including promoter activation, splice-switching oligonucleotides, and the stabilization of SMN transcript and/or protein [25, 47–49]. Recent clinical success with the approved SPINRAZA have shown that splice switching oligonucleotides is indeed efficient, achieving elevated SMN levels and robust phenotypic corrections. Yet, this drug is a new chemical entity and may or may not fail post-marketing. Recent examples of recalled FDA-approved drugs include VIOXX (rofecoxib), MERIDIA (sibutramine) and others. Thus, despite SPINRAZA approval, decades-familiar drugs have a major advantage, either as monotherapy or in combined therapy in which two different mechanisms of actions are exploited.
The repurposing pharmaceutical approach was indeed sought after in SMA treatment as many repurposed drugs were researched in the past, even in advanced clinical trials, including [1] histone deacetylase inhibitors (HDACs), hydroxyurea, and prolactin for the activation of SMN2 gene expression; [2] salbutamol for splicing modulation; [3] aminoglycosides for protein translation; and [4] riluzole and ceftriaxone for neuroprotection [50]. Regretfully, none of those drugs was approved for SMA therapy. Recently, another repurposed drug, the macrolide antibiotics azithromycin (AZM), was shown to enable read through of the SMN2 gene [24], compensating SMN protein levels in patients derived cell lines. It was not known, however, if AZM was able to treat SMA mice models and correct the severe SMA phenotype.
As demonstrated in this report, AZM can be administered to the CNS as a monotherapy, resulting in modest, but statistically significant improvements in several aspects of the disease, yet not extending survival. However, AZM’s activity as an adjunctive therapy with the splice-switching ASO allowed the use of relatively low concentrations of both therapies, thus potentially improving the safety profiles of both. The importance of these observations is directly related to clinical applicability: 1) AZM is FDA approved and could be moved quickly into SMA patients; 2) AZM can induce SMN protein in disease relevant tissues; 3) AZM has attractive pharmacodynamics as it is relatively stable and was shown to retain activity for extended periods of time (unpublished data); 4) intrathecal administration of AZM at low levels would be efficacious in disease affected tissues without reaching the blood and affecting other tissues such as the gastrointestinal tract (unpublished data); and [5] AZM can be administered at the same route and schedule as SPINRAZA, a plausible candidate for combinatorial therapy.
Low levels of the SMN protein cause SMA, hence, restoring SMN has been the major focus of developing therapeutic strategies and testing them in clinical trials. However, having two distinct pathways that address different components of the disease or induce SMN through two distinct mechanisms are intriguing therapeutic combinations. Plastin-3 (PLS3) has been shown to be a modifier of the SMA phenotype. Interestingly, re-introduction of PLS3 either genetically or with an AAV9 vector, does not increase SMN, demonstrating that an alternative functional pathway is responsible for the improved phenotype [51, 52]. Recently, dual-therapeutic approaches were shown to produce an enhanced improvement when PLS3 and a splice-switching ASO were administered to severe SMA animals [53, 54]. Both groups have reported that PLS3 co-administration improves components of the axonal phenotype and extends survival in a pharmacologically induced model of SMA when administered with a splice-switching ASO that increases SMN production. In a similar vein, co-administration of a SMN2 exon 7 splice modifying small molecule and AAV1-follistatin resulted in enhanced muscle pathology [55]. As follistatin is a potent stimulator of skeletal muscle growth and development by inhibiting myostatin, these results provide further evidence that combinatorial cocktails can be an important tool going forward. However, it is likely that these cocktails will need to address important issues such as: 1) improving the underlying SMN deficiency; and 2) provide functional SMN-independent support through disease-relevant pathways, such as neuroprotection, skeletal muscle enhancement, cellular trafficking (PLS3-mediated activity), actin dynamics, and perhaps many others.
With the recent FDA approval of the first SMA therapeutic, SPINRAZA, there has been tremendous excitement in the SMA community. This opens up the possibility of moving forward with adjunctive therapies that function through different modes of action, either to increase SMN or to provide a distinct function such as neuroprotection or skeletal muscle enhancement. Whether this type of strategy will increase efficacy and safety or broaden the patient population that responds to particular therapies is an important area of research to address in the immediate future.
FUNDING
This work was funded by grants from BioBlast Pharma Ltd.
CONFLICT OF INTEREST
CLL is co-founder and Chief Scientific Officer of Shift Pharmaceuticals. DM and HG are employees of BioBlast Pharma Ltd.