
Abstract
Sleep and circadian disruption affects most individuals with Huntington’s disease (HD) at some stage in their lives. Sleep and circadian dysregulation are also present in many mouse and the sheep models of HD. Here I review evidence for sleep and/or circadian dysfunction in HD transgenic animal models and discuss two key questions: 1) How relevant are such findings to people with HD, and 2) Whether or not therapeutic interventions that ameliorate deficits in animal models of HD might translate to meaningful therapies for people with HD.
INTRODUCTION
Sleep disorders seen in HD may arise from either dysfunction of the pathways that regulate sleep and wake or from circadian dysfunction, since the sleep/wake cycle is a circadian-driven behavior. The evidence for sleep and circadian dysfunction in people with HD is reviewed in this special issue by Saade-Lemus and Videnovic [1] (for other reviews, see [2–5]).
The primary symptom of circadian rhythm disorder in humans is an inability to sleep at the desired time. This may be reflected by excessive daytime sleepiness. There may also be a shift of day/night activity patterns. As well as behavioral changes, disturbed circadian rhythmicity may cause disruption of rhythms of hormones such as melatonin, cortisol, and prolactin. Furthermore, internal misalignment of the body clock and the sleep-wake cycle may result in disturbed sleep electroencephalogram (EEG).
ANIMAL MODELS OF HUNTINGTON’S DISEASE
There are numerous transgenic mouse models of HD. These include models carrying only a fragment of the mutant human HTT gene (R6/1 and R6/2), artificial chromosome models (BAC HD and YAC 128), and full length knockin models (zQ175 and the Hdh series). For reviews, see [6–8]. As well, there is a transgenic HD rat [9], and several transgenic HD large animal models including two pig [10, 11], one sheep [12], and three non-human primate models (for references, see [13]). There are also invertebrate models of HD. To date, however, no studies of sleep or circadian rhythms have been conducted in any non-human primate models, and although behavioral correlates of sleep have been noted in invertebrates, they do not have mammalian-like sleep pathways, and will not be discussed here. Clear circadian behavioral and/or biochemical/gene expression abnormalities have been measured or described in mouse, rat, pig, and sheep models, so in this review I will focus on the results of studies in these species.
Studying sleep and circadian rhythms in mouse models comes with caveats. Sleep can be measured directly in humans using EEG and sleep-wake EEG activity correlates broadly with motor activity in humans. The EEG in humans is generated predominantly by activity in the cortex. EEG is less easy to study in rodents since surgical implantation of electrodes is required. Furthermore, the EEG activity in rodent brain is dominated by theta activity generated from the hippocampus. By contrast, locomotor activity in rodents easily measured and is commonly used as a behavioral ‘readout’ for sleep. The major pathways for sleep and circadian rhythmic behavior are present in rodents (see review by Nollet et al., this issue [14]), as is the intrinsic cellular circadian clock machinery (see review by Patton and Hastings, this issue [15]). Thus, rodents make excellent models for studying sleep and particularly circadian rhythms. Having said that, there are several challenges in translating data from humans to rodent models, and vice versa, that need to be considered when comparisons are made. For example, mice and rats are nocturnal under laboratory lighting conditions, whereas humans are diurnal. As well, circadian rhythms in humans are modulated by melatonin, and onset of sleep is regulated by a dim light onset of a surge in melatonin. Most laboratory mouse lines, including the commonly used C57BL/6 strains, however, do not make melatonin. It is not clear how much this matters, but the translational reliability of melatonin studies in mice has been questioned [16, 17]. Another issue is the disease-causing mutation in the HTT gene itself. The HD mutation in humans is an unstable CAG repeat that is translated to a polyglutamine repeat in exon 1 of the huntingtin protein (HTT). A major challenge for translational studies is that while numerous HD mouse models exist, the gene context of the mutation they carry is not the same as it is in humans. First, the CAG repeat mutation that gives rise to an overt behavioral phenotype in mice is much longer that that seen in people with HD (>150 CAGs in mice, whereas most people with HD have a repeat length of 40–50). Second, many of the mouse lines used for circadian studies carry the mutation in a fragment of the HTT gene (R6/1, R6/2). Third, the CAG repeat mutation is unstable in the mutant HTT gene and the instability depends on whether or not the CAG repeat is ‘pure’ or interrupted by CAA (that also codes for glutamine). BAC and YAC HD mice both have interrupted repeats and do not show instability. The CAG repeat in R6/2 mice is pure CAG and unstable. Unfortunately, the number of CAG repeats is not always reported. Since the age at onset and rate of progression depends upon the CAG repeat length, unless the strain and the CAG repeat length is similar, it is not possible to compare data directly between laboratories. Nevertheless, despite these caveats, data emerging from studies using mouse models suggest at multiple levels that there are both sleep and circadian abnormalities arising in mice carrying the HD gene.
CIRCADIAN RHYTHM DISTURBANCE IN HUNTINGTON’S DISEASE MODELS
Mice
The first hint that circadian biology is disturbed in HD came from a study showing that an experimental pharmacological treatment that improved cognitive decline in R6/2 mice reversed the abnormal expression of multiple genes that were dysregulated in HD [18] These included the family of ‘circadian rhythm’ genes. This was not only the first evidence linking circadian dysfunction and HD, but also the first study showing that cognitive decline might be linked to circadian dysfunction in HD mice. A follow-on study in which circadian behavior was measured directly found major disruption of circadian rhythmicity in the R6/2 mice [19]. Using passive infrared sensors in a climate- and light-controlled cabinet environment to measure circadian behavior around the clock, night-day behaviors of R6/2 and wildtype (WT) littermate control mice were shown to be similarly normal until they were about 8 weeks of age, with free-running (endogenous) rhythms of around 24 h as expected. After that, the pattern of activity of R6/2 mice changed, with more activity during the day and less at night compared to WT mice. Daytime activity increased and nocturnal activity fell in the R6/2 mice, eventually leading to the complete disintegration of circadian behavior. By 16 weeks R6/2 mice had no endogenous circadian rhythm at all (Fig. 1). An age-dependent disruption in behavioral rhythms has now been documented in all the HD models in which it has been studied, including R6/2; R6/1, BACHD, Q140, Q175, and YAC128 lines [19–31].
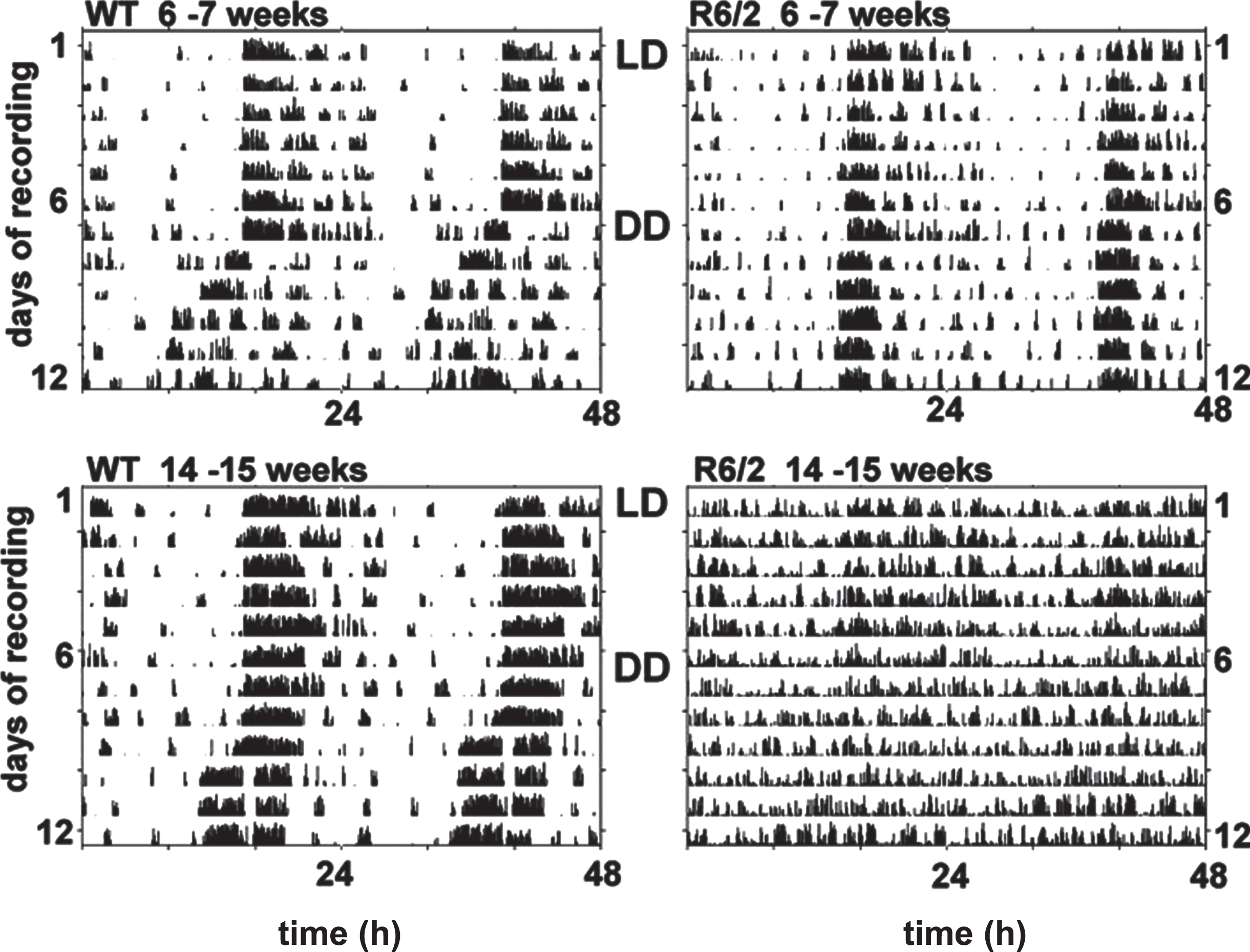
Disruption of circadian activity patterns in R6/2 mice with age. Typical double-plotted actograms show continuous recordings of a WT (left) and R6/2 (right) mice monitored for 1 week under LD light conditions (12 h light and 12 h dark) from 6-7 weeks (top panels) or 14-15 weeks (lower panels) of age. Mice were transferred into DD at 7 or 15 weeks of age, and their activity was recorded for an additional 7 d. This figure is an adapted version of Fig. 2 in Morton et al. [19].
In R6/2 mice the disintegration of circadian rhythms was accompanied by marked disruption of clock genes mPER2 and mBmal1 in the central circadian clock, the suprachiasmatic nucleus (SCN) of the thalamus as well as motor cortex and striatum [19]. A subsequent study showed that peripheral clocks were also dysregulated [32]. At a molecular level, alterations in the amplitude or phase of the rhythms in core circadian clock gene expression were seen in several brain regions beyond the SCN, including striatum, hippocampus, and motor cortex [19] and peripheral tissues, particularly the liver [32, 33]. Notably, pathophysiology is also described in HD mouse SCN [34].
Interestingly, the dysfunction in circadian clock function may not be cell-autonomous. In a study exploring SCN deficits at a circuitry level, electrophysiological recordings made from the SCN showed that despite the strong behavioral deficits, the SCN was intrinsically competent in the R6/2 mice [35]. It was concluded that the circadian dysfunction arose from abnormalities in brain circuitry afferent to the SCN rather than a primary deficiency in the SCN. Indeed, where explants of the SCN were used, although clock genes were strongly dysregulated in mice with disintegrated behaviors, SCN explants from those same animals recovered their rhythms within a day or so [19]. Thus the clock machinery appeared to be intact, although its regulation was severely disrupted. This evidence supports the idea that circadian dysfunction is driven by system-wide influences on clock gene expression rather than impaired intrinsic function at a cellular level due to mutant huntingtin (mHTT).
In summary, there is now extensive evidence supporting the idea that circadian disruption is found at physiological levels in HD rodent models. Not only abnormal day-night activity deficits are seen (as detailed above), but also deficits in other rhythms controlled by the SCN, including core body temperature [24], sleep [36–38], circadian controlled cardiac functions including heart rate variability [24, 39–42], and endocrine function [43, 44] are seen in many models. Most recently, circadian-regulated metabolites have been shown to be abnormal in R6/2 mice [45].
There are two additional circadian oscillators that are independent of the SCN. The first is the food-entrainable oscillator (for more see below), the second is a methamphetamine-sensitive circadian oscillator (MASCO). The MASCO is distinct from canonical circadian oscillators because it controls robust activity rhythms by mechanism that are independent of the suprachiasmatic nucleus and circadian genes are not essential for its timekeeping. There is a major dysfunction of the MASCO in presymptomatic R6/2 mice that is likely to be due to an early abnormality of the catecholaminergic systems [46]. A similar deficit is seen in the Q175 full length knockin mouse [47]. In R6/2 mice the deficit can be restored by treatment with the antidepressant paroxetine [48]. This oscillator has not been studied in any other HD model, but the findings of progressive deficits in two HD models should be of interest, particularly given that a proposed function of MASCO is to regulate sleep-wake cycles [49].
HD sheep
The transgenic sheep model of HD carries a full length human cDNA with a CAG repeat of 73 [12]. This model has measurable phenotype in multiple domains but has rarely been used. This is likely to be due to several factors, not least the fact that access is geographically restricted since the stock flock of these sheep is in Australia. Sheep are not a standard laboratory animal, although many research facilities have capacity to keep large animals. But the infrequent use of a promising model may also be because the HD sheep shows no overt signs of disease for at least 10 years. While this is consistent with the CAG mutation length that would be expected to manifest in humans at a juvenile age, it is also a timeframe that few researchers/funders are prepared to entertain. Nevertheless, given that even at a presymptomatic age, the HD sheep shows relevant pathological, behavioral, metabolic, and physiological phenotypes, it is an extremely interesting model for studying circadian and sleep in HD (for references, see [50]). Notably, the sheep is also the only large animal model of HD in which sleep EEGs have been recorded and significant sleep abnormalities are present even at the early stage of disease (for details, see Sleep section below).
Major differences between normal and HD sheep have been observed in circadian behavior at 3 and 5 years [51] and in melatonin secretion at 5 and 7 years [52]. Circadian behavior in HD and normal sheep was measured using GPS tracking devices [51]. Sheep activity was measured around the clock for several months in groups of sheep that were either single genotype or mixed. Sheep were aged either 3 or 5 years, i.e., at least 5 years before the onset of symptoms. Compared to control sheep, HD sheep showed clear abnormal patterns of activity that worsened with age. In particular, the HD sheep showed increased evening activity and delays in ‘settling’ down to sleep for the night, behavior that was reminiscent of the early evening behavioral disturbance known as ‘sundowning’ that is seen in Alzheimer’s disease patients (for references, see [53]). Of particular interest is that while there were pronounced differences between normal and HD sheep when they were kept in single genotype flocks, circadian behavior abnormalities were ameliorated when HD and control sheep were housed together. The fact that HD sheep responded normally to circadian cues when housed with normal sheep suggest that at least part of the circadian dysfunction in HD sheep is due to altered responsiveness of HD sheep to extrinsic Zeitgebers, which in this case were probably social cues.
In a recent metabolomics study, Spick et al. found both absolute and rhythmic differences in metabolites in HD at 5 and 7 years compared to age-matched control sheep [54]. An increase in both the number of disturbed metabolites and the magnitude of change of acrophase (the time at which the rhythms peak) was seen in samples from 7-year-old HD compared to control sheep. There were striking similarities between the dysregulated metabolites identified in HD sheep and human patients (notably of phosphatidylcholines, amino acids, urea, and threonine).
Circadian changes in other species
While few studies have been conducted using other species, these also hint at circadian dysregulation. Research using the tgHD rat showed hyperactivity during the dark cycle and more frequent activity during the light cycle [22], suggesting circadian dysregulation. In the transgenic HD pig, one study showed that there is more frequent rooting activity in HD minipigs than in wildtype minipigs [55] that was consistent with a result indicating that during the daytime, transgenic HD minipigs rested less than wildtype minipigs [10].
MEASURING CIRCADIAN BIOMARKERS IN ANIMAL MODELS
Melatonin
One of the best-understood circadian circuits is the temporal control of melatonin secretion. The circuits involved in the nightly rise in melatonin secretion and autonomic function are well understood [56]. The SCN expresses high levels of melatonin receptors, and the interaction between the SCN and pineal gland forms a neuro-endocrine feedback loop [57]. Dim light melatonin onset (DLMO) is used as a laboratory measure to determine the phase of the circadian cycle in humans [58]. In humans and sheep melatonin levels can be measured in plasma or saliva. ‘Around-the-clock’ measurements of melatonin allow precise determination of phase. As mentioned above, the impact of HD pathology on melatonin rhythmicity cannot be studied in mouse strains that do not produce melatonin.
Because they make and secrete melatonin and are large enough to enable serial blood samples to be collected easily, it has been possible to study melatonin secretion around-the-clock in HD sheep. Melatonin secretion is abnormal in presymptomatic HD sheep [52] and changes progressively. The direction of change, however, is opposite to what would be expected, with increased melatonin secretion seen in 5-year-old HD sheep compared to control sheep. At 7 years of age, this elevated melatonin is reduced. This contrasts to studies of melatonin secretion in HD, where either a delayed phase [59] or reduced melatonin levels were reported in people carrying the mutant HTT gene [60]. Clearly more work is needed in this area to determine if the differences between human and sheep are due to species differences or to the stage of disease. The HD sheep in this study were at a presymptomatic age, and no study of symptomatic sheep has yet been conducted. It will be interesting to see if closer to symptomatic onset the melatonin level in HD sheep fall below those of normal sheep as would be predicted from the human studies.
MEASURING SLEEP IN HUNTINGTON’S DISEASE ANIMAL MODELS
The EEG has long been a valuable tool for diagnosing sleep abnormalities. Quantitative spectral analysis is one of the standard methods used for quantification of the EEG, with the power spectrum reflecting the distribution of signal power over frequency. EEG studies in HD patients show loss of theta (4–7 Hz) power in REM and NREM sleep [61, 62] and an increase in gamma (25–40 Hz) content of NREM sleep [62]. While quantitative EEG is the most accurate way to study sleep in humans, there is a paucity of direct evidence of abnormalities in sleep EEG in people with HD, and those data available are not consistent between studies (for references, see [63]). This, however, is likely to reflect the challenges of studying EEG in HD patients rather than the lack of abnormalities.
HD mice
There is comprehensive evidence of abnormal EEG-defined sleep architecture in mouse [36–38, 64–66]. Importantly, these studies followed alterations in EEG-defined sleep longitudinally, with sleep EEG recorded regularly together with a wide range of behavioral tests to follow the age-dependent progression of the phenotype. These studies consistently found an early and progressive deterioration of both sleep architecture and behavior in HD mice. In addition, they found alterations in the EEG power spectrum including a reduction in delta (1–4 Hz), a peak frequency shift of theta rhythm from 7 Hz to 6 Hz during rapid eye movement sleep, as well as abnormal beta power [68], state-dependent increases in power of gamma oscillation that were reminiscent of those seen in HD patients [62] and also schizophrenic patients during sleep and events of psychosis [69, 70].
HD sheep
There is clear evidence of abnormal sleep EEG in HD sheep [63, 68]. In fact, the nature of the abnormalities seen in the sheep studies may in part explain the lack of clear changes reported in early-stage HD. Schneider et al. examined the number of transitions between sleep and wake were similar in normal and HD sheep. They found that in the HD sheep, however, while the number of transitions were similar, the dynamics of transitions from sleep-to-wake differed markedly between genotypes. Rather than the gradual changes in EEG power that occurs during transitioning from sleep-to-wake in normal sheep, transition into the wake state was abrupt in HD sheep [68].
In a separate study using the same sheep, quantitative parameters (amount of time spent in rapid eye movement sleep (REM) and non-REM sleep) were again similar between genotypes. Major differences, however, were seen between HD and normal sheep in both EEG power and its pattern of distribution during non-rapid eye movement sleep (Fig. 2A). In particular, the distribution of oscillations that typically dominate the EEG in the first half of the night of normal sheep were skewed in the transgenic sheep to be dominant in the second half of the night. In this study the effect of sleep deprivation was also compared in normal and HD sheep. Interestingly, disruption of sleep in normal sheep caused a change the pattern of distribution of EEG powers so they looked more like those seen in HD sheep under baseline conditions. Thus, it is possible that transgenic HD sheep exist in a state that resemble a chronic state of physiological sleep deprivation. Furthermore, homeostatic sleep rebound was also abnormal in HD sheep compared to normal sheep (Fig. 2B) [63]. Notably the pattern of changes we saw in the HD sheep, that show no overt signs of disease and whose amount of sleep was similar to the normal sheep, would not have been detected using standard quantitative measures of sleep such as amount of NREM, REM and wake. There have been no studies of sleep deprivation done using people with HD as subjects. Extrapolating from the HD sheep EEG sleep data, it is possible that in HD, particularly at early stages of disease, while quantitatively sleep may seems normal, functionally it may be abnormal. This study emphasizes the need to not only measure amount of sleep, but also the quality and distribution of sleep across the night.
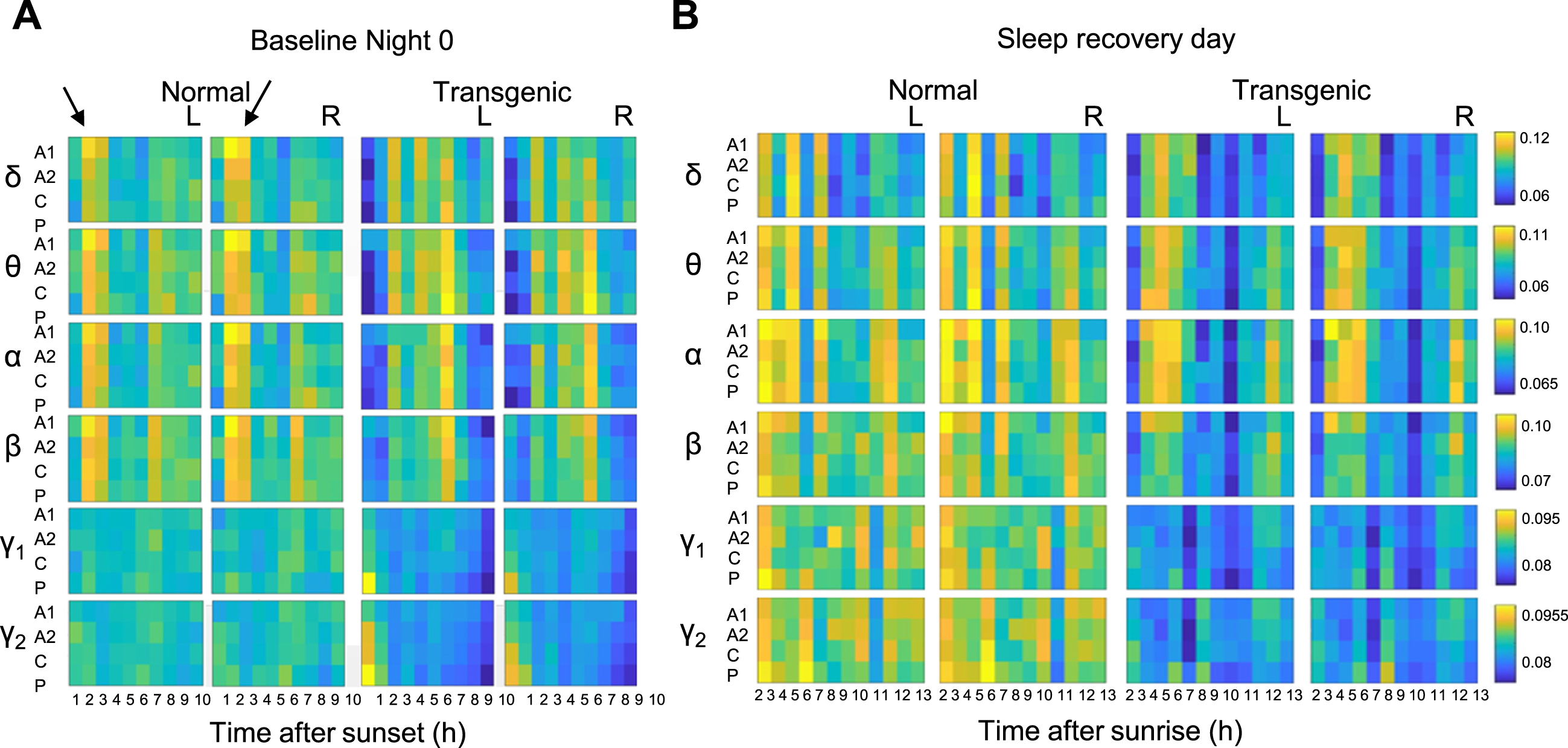
EEG powers during non-rapid eye movement sleep are different in transgenic and normal sheep under baseline conditions and after sleep deprivation. Heatmaps show the relative distribution of EEG powers in different frequency ranges denoted by Greek letters (δ, delta [0.5–4 Hz]; θ, theta [4–9 Hz]; α, alpha [9–14 Hz]; β, beta [14–35 Hz]; γ1, low-gamma [35–55 Hz]; γ2, high-gamma [55–120 Hz]) during an undisturbed light-off (Baseline Night 0; A) and on the day following a night of sleep deprivation (B) in transgenic and normal sheep. The arrows on A point to the period when the distribution of EEG powers show striking difference between normal and transgenic sheep at night. Color scales in each frequency range show the mean normalized EEG power values (per channel, per hour). For full results and experimental details see [63]. L and R = left or right hemisphere respectively. This figure is an adapted version of Figs. 5 and 7 in Vas et al. [63].
Other models
To my knowledge, there have been no EEG studies conducted using HD rats or HD pigs.
THE RELATIONSHIP BETWEEN SLEEP DISTURBANCE AND OTHER HUNTINGTON’S DISEASE SYMPTOMS
The symptoms of HD include abnormalities in motor control, cognitive deficits (particularly attention and executive function as well as impairments in memory and learning) and psychiatric symptoms such as low mood and depression. Sleep and circadian disturbances are an increasingly recognized as a feature of HD and may be seen in up to 90% of people with HD [71]. There is likely to be a link between cognitive/psychiatric symptoms and sleep disturbances, but this is one aspect that is difficult to study in animal models. There have been no studies of cognitive or psychiatric symptoms in sheep or pig models, and although there are correlates in mice for depression anxiety and cognitive decline in mouse models of HD, effects of sleep disturbance on ‘affect’ have been deduced rather than shown directly.
There is evidence suggesting the existence of a bidirectional feedforward cycle between sleep disturbances and neurodegeneration. In rats and mice, sleep deprivation has been shown to induce oxidative stress, neuroinflammation [72], and aberrant protein homeostasis [73]. Furthermore, the discovery that the glymphatic system (that is thought to play a critical role in the clearance of misfolded proteins from the brain) is more active during slow wave sleep [74] has led to the idea sleep deprivation may reduce misfolded protein clearance and potentially contribute to disease progression. There is evidence for this in both Alzheimer’s [75] and Parkinson’s diseases [76, 77]. For HD there is, as yet, no evidence. Interestingly, however, a recent study shows that pathological hallmarks of HD (somatic CAG repeat instability and abnormal protein aggregation) are best correlated with early-onset striatum-selective molecular pathogenesis, locomotor dysfunction, and sleep deficits [30].
CAUSE OR CONSEQUENCE: COGNITIVE ABNORMALITIES AND SLEEP DYSFUNCTION
HD mice
There is both indirect [18] and direct [78–80] evidence that cognitive deficits and circadian dysfunction are linked. Pharmacological imposition of sleep and wakefulness using alprazolam and modafinil markedly improved cognitive performance in a two-choice visual discrimination task in the R6/2 line of transgenic HD mice [78, 79] and reversed some EEG spectral changes [81]. Treatment with the orexin receptor antagonist suvorexant was found to alleviate cognitive deficits in R6/1 mice [80]. Unexpectedly, in the study by Pallier and Morton, the treatment of HD mice housed in the same cage as vehicle-treated mice also improved the cognitive function of those mice. This is likely to be driven by social cues from the treated mice with improved sleep/circadian patterns. The importance of social cues cannot be over emphasized. Not only might positive social cues improve sleep in people with HD, but negative cues may also have the opposite effect. This has not been studied with respect to HD, but it has been seen in normal controls subjects. In an early study of circadian behavior in people with HD, two control groups were used, one that comprised the neurologically normal carers of the HD subjects, the other comprising a non-carer group. Notably, sleep in the carer group was significantly worse than that of the non-carer group and not different from the HD patient group.
BEHAVIORAL INTERVENTIONS IN HD MOUSE MODELS
A number of non-pharmacological interventions the affect circadian rhythms have been tested in HD mouse models, in particular, environmental enrichment (EE), and changes to light and scheduled feeding.
Changes to light schedules
Light is an important Zeitgeber with the circadian timing system synchronized by the light/dark cycle. Changing environmental lighting is a robust way to regulate circadian rhythms and possibly influence the sleep/wake cycle. A detailed discussion of the pathways controlling the effect of light on circadian rhythms is beyond the scope of this review but has been well described elsewhere [82–84]. R6/2 mice have retinal abnormalities in ipRGCs [85, 86] but respond abnormally to photic entrainment even before they have retinal abnormalities in ipRGCs [87]. Nevertheless, two studies have demonstrated that changes in light exposure benefit HD mice. In the R6/2 model a long-day photoperiod (16 h light/8 h dark cycle; 100 lux) improved some aspects of daily locomotor rhythms and significantly improved the survival of R6/2 female mice compared to mice kept under standard conditions (12 h light/12 h dark LD cycle [88]. In another study, light therapy designed to strengthen the intrinsic circadian timing system delayed disease progression in BACHD and Q175 mice [89]. After treatment with 6 h of blue light at the beginning of their daily light cycle for 3 months, both WT and HD mice showed improvements in their locomotor activity rhythm without significant change to their sleep behavior. Treated HD mice of both lines exhibited improved motor performance compared to untreated HD mice. This work could be translated easily to people with HD, although one caveat is that intrinsically photosensitive retinal ganglion cells (that express the photopigment melanopsin and control circadian response to light) degenerate in HD mice [86, 87]. It is not known if similar changes occur in HD patient retina.
Environmental enrichment
EE is not a Zeitgeber per se, but applying EE during the dark when mice are normally active, enhances activity levels and conversely, applying EE during the normal rest time may have the effect of weakening rhythms and may also impose a sleep-deprivation effect. A variety of types of EE have been shown to have a positive effect on the progression of motor symptoms and the survival of the R6/1 and R6/2 mouse models of HD [90–96]. Beneficial effects became apparent after as little as 2 weeks [95]. EE improves neurological function and cognitive performance in animal models of neurodegenerative disease including HD [97].
Scheduled feeding
Timing of feeding is an important regulator of the circadian system [98], and there is good evidence for an SCN-independent oscillator that has been named the ‘food-entrainable’ oscillator [99–101]. Early work in the R6/2 model found HD-driven disruption in circadian gene expression in the liver [32] and circadian rhythms of clock-driven genes that are key metabolic outputs in the liver were abolished in vivo. Importantly, at least some of these deficits were corrected by a feeding schedule that allowed access to food 5 h per day, with time of feeding deliberately selected to occur when the mice would normally be sleeping (ZT 6–11), ensuring that any restoration of liver gene expression could be attributed directly to the feeding schedule. In a later study using the R6/2 mouse, food entrainment was imposed by allowing access to food only during the dark period. This feeding schedule slowed the loss of body weight, aided the maintenance of body temperature and improved locomotor behavior [102]. The effect of timed restriction of food has also been studied in the BACHD mouse model [103] and Q175 models [104], where mice were exposed to a feeding period (6 h) aligned with the middle of the period when mice are normally active. Mice on this restricted feeding regime showed improvements in their locomotor activity and sleep behavioral rhythms and heart rate variability, with motor improvements correlated with improved circadian output. The mechanisms underlying these benefits of time restricted feeding are not known, although improvements in metabolism that occur in HD would seem to be a logical line of investigation, since metabolism is dysregulated in HD models [32, 54].
Together these studies support the idea that strengthening the circadian system may delay the progression of disease in HD mouse models.
PHARMACOLOGICAL MODULATION OF SLEEP/CIRCADIAN DISORDER IN HD MICE
Hypnotics and antidepressants
There are numerous pharmaceutical agents available on prescription for insomnia and other sleep disorders (for a review, see [105]). Given that there has never been a systematic study of the therapeutic efficacy of hypnotics on sleep in people with HD, sleep disorders associated with HD are treated empirically (see review by Owen et al., this issue [106]). The safety of these drugs had been established for short-term treatment in normal subjects, but more research is needed into their long-term effectiveness in treating insomnia in HD.
There are some studies using HD mice that suggest that systematic use of hypnotics may have beneficial effects in HD. For example, daily treatment with a short-acting benzodiazepine (alprazolam) [79] improved cognitive performance of R6/2 mice and normalized the dysregulated expression of the core clock gene Period2 and the neuropeptide Prokineticin 2. In a follow up study [78], a combination of alprazolam (given before the rest period to improve sleep) and the stimulant modafinil (given at onset of the active period) was used in R6/2 mice. Both drugs alone improved cognitive function and reduced apathy but had a stronger effect when used in combination. In R6/2 mice, both zolpidem and the tricyclic antidepressant amitriptyline suppressed the abnormally high REM sleep amount and EEG gamma (30–46 Hz) oscillations in a dose-dependent manner [107, 108]. The effect of amitriptyline on sleep was similar in both R6/2 and WT mice while zolpidem was effective in WT but not in R6/2 mice. This finding highlights the importance of examining directly the effect of drugs on HD mice and not assuming that a drug that works in a WT mouse or person will have the same effect in an HD animal or a person with HD. The serotonin-reuptake inhibitors paroxetine was shown to improve circadian parameters in the mouse model [48]. Furthermore, administration of suvorexant (orexin receptor 1 and 2 antagonist) improved sleep in R6/1 mice [79]. This interesting study showed a 5-day treatment with suvorexant improved body weight and spatial recognition memory without altering rotarod performance in female mice. Unfortunately, this study did not evaluate whether rhythms in activity or sleep were improved by suvorexant.
Histamine is one of the main transmitters that control arousal [109], and it is a potent regulator of the circadian system [110, 111]. Histamine release is circadian controlled, and histamine-3 receptor (H3R) is a promising target to treat hypersomnia and H3R antagonists promote wakefulness. One study using a H3R antagonist (GSK189254) administered before the rest period for 4 weeks in Q175 mice found strengthened activity rhythms, improved cognitive performance, and improved mood (as measured by the tail suspension test) [112]. It also reduced inappropriate locomotor activity during the normal sleep time.
Melatonin
No systematic studies of the use of melatonin have been conducted in either HD mice or sheep. Furthermore, although melatonin secretion is abnormal in presymptomatic HD sheep [52] and changes progressively, the direction of change is opposite to what would be expected (see above). Clearly more work is needed in this area. Our lack of knowledge about the impact of HD pathology on melatonin rhythmicity is complicated by the fact that many mouse models are on the C57BL/6 background that do not produce melatonin.
Summary of selected animal models of Huntington’s disease
ND, not done; NHP, non-human primate; EEG, electroencephalogram; BAC, bacterial artificial chromosome; YAC, yeast artificial chromosome; ?, possibly. For further references see. 1Farshim PP, Bates GP. Mouse models of Huntington’s disease. In: Precious S, Rosser A, Dunnett S, editors. (eds) Huntington’s Disease. Methods in Molecular Biology, vol 1780. New York: Humana Press; 2018. p. 97–120. doi: 10.1007/978-1-4939-7825-0_6. 2Chiem E, Zhao K, Stark G, Ghiani CA, Colwell CS, Paul KN. Sex differences in sleep phenotypes in the BACHD mouse model of Huntington’s disease. bioRxiv [Preprint]. 2023 Apr 28:2023.04.28.538324. doi: 10.1101/2023.04.28.538324. 3Manfré G, Clemensson EKH, Kyriakou EI, Clemensson LE, van der Harst JE, Homberg JR, et al. The BACHD rat model of Huntington disease shows specific deficits in a test battery of motor function. Front Behav Neurosci. 2017;11:218. doi: 10.3389/fnbeh.2017.00218. 4Howland D, Ellederova Z, Aronin N, Fernau D, Gallagher J, Taylor A, et al. Large animal models of Huntington’s disease: what we have learned and where we need to go next. J Huntingtons Dis. 2020;9(3):201-16. doi: 10.3233/JHD-200425.
WHERE TO GO FROM HERE?
The HD field is ideally positioned to study therapeutic avenues for the treatment of sleep and circadian disorder in neurological diseases [113]. Numerous mouse models exist, and while none recapitulate the human condition exactly, nevertheless there is already enough evidence to show that they have both sleep and circadian deficits that are relevant to the human condition. Importantly, circadian biology in mice is well understood at both a behavioral and molecular level. Several drug studies show that circadian and sleep deficits can be effectively treated or prevented. Better use of mice should be made to explore potential treatments. Similarly, the HD sheep is an underused resource. To date, no studies of interventional therapies have been conducted using the HD sheep. Nevertheless, given that they have clear EEG abnormalities, sheep are ideal for studying mechanisms underlying sleep dysfunction and the relationship to cognitive behavior in HD. Furthermore, sheep could be used to study the effects of sleep deprivation in HD. Since during the sleep recovery period, normal sheep showed a significant ‘rebound’ increase in delta power with frontal dominance, but a similar rebound was not seen in transgenic sheep it seems likely that their homeostatic response to sleep deprivation is abnormal. Although sleep abnormalities in early-stage HD are subtle, with patients often unaware of their existence, they may contribute to impairment of neurological function that herald the onset of disease [114]. A better understanding of the mechanisms underlying EEG abnormalities in early-stage HD would give insight into how, and when, they progress into a clear sleep disorder in humans.
The possibilities of sleep and circadian studies in the HD sheep are compelling for two reasons. First, the HD sheep up to 10 years of age, are presymptomatic. Nevertheless, significant changes in organization of sleep have been measured (see above). Secondly, studies of metabolism and hormone-related parameters are very difficult to control in humans, where different food, different work/home lifestyles may all add variability to measure parameters. By contrast, many of the parameters that sleep scientists would like to control but find difficult in humans, can be controlled well in sheep. Sheep are thus ideal subject for sleep and circadian studies, since they can be habituated to a single environment, fed the same diet, placed under the same light cycle, and blood samples taken around the clock so their blood samples could be assessed directly but also with respect to dim light melatonin onset. It is not possible to do this in groups of people.
It is difficult to disentangle the circadian clock system and sleep/wake cycle as they are multi-layered and heavily interconnected systems. While indirect evidence favors circadian dysfunction being a primary cause of sleep disruptions, evidence from qEEG in people with HD [62], mice [36–38, 108], and sheep [63, 68] suggests there are may also be underlying abnormalities in sleep EEG generation. More studies of both circadian and sleep physiology should be conducted in HD animal models.
Although few studies of people with HD include detailed quantitative EEG analysis, given the concordance of evidence from HD humans, sheep, and mice in terms of the changes in EEG power, future studies in people with HD should include qEEG, particularly since disease-driven changes in EEG may serve as a biomarker for disease progression in HD [115].
CONCLUSIONS
Sleep is fundamentally important to human health and subjective experience of wellness. There is unequivocal evidence that HD mice have abnormal circadian control of sleep, and growing evidence that this may also be the case in people with HD. There are numerous lines of evidence in normal people that sleep and circadian disturbances are linked to cognitive deficits and mood disorders. Logically, it seems likely that sleep and circadian disturbances would exacerbate cognitive deficits and mood stability in HD. Indeed, HD patients, who have impaired neurological function, may be particularly vulnerable to the deleterious effects of sleep and circadian disturbances. It is possible that sleep disturbances may contribute to HD symptoms and may exacerbate cognitive deficits. More basic research is needed to understand the impact of sleep and circadian abnormalities in people with HD. Studies in HD animals (both those demonstrating that there are sleep and circadian deficits and those showing that these deficits are not hard-wired but can be reversed with behavioral therapies or pharmaceutical agents) provide compelling evidence that this should be better studied in humans. There are numerous therapies aimed at improving sleep/circadian disorders using drugs that are already approved for use in humans. Regardless of the exact role sleep disturbances play in HD pathology and its symptoms, the negative effect on patient and carer is indisputable. This should be reason enough to prompt further research and strive to find treatments.
CONFLICT OF INTEREST
The author has no conflicts of interest to report.