
Abstract
Ischemic stroke causes secondary neurodegeneration in the thalamus ipsilateral to the infarction site and impedes neurological recovery. Axonal degeneration of thalamocortical fibers and autophagy overactivation are involved in thalamic neurodegeneration after ischemic stroke. However, the molecular mechanisms underlying thalamic neurodegeneration remain unclear. Sterile /Armadillo/Toll-Interleukin receptor homology domain protein (SARM1) can induce Wallerian degeneration. Herein, we aimed to investigate the role of SARM1 in thalamic neurodegeneration and autophagy activation after photothrombotic infarction. Neurological deficits measured using modified neurological severity scores and adhesive-removal test were ameliorated in Sarm1−/− mice after photothrombotic infarction. Compared with wild-type mice, Sarm1−/− mice exhibited unaltered infarct volume; however, there were markedly reduced neuronal death and gliosis in the ipsilateral thalamus. In parallel, autophagy activation was attenuated in the thalamus of Sarm1−/− mice after cerebral infarction. Thalamic Sarm1 re-expression in Sarm1−/− mice increased thalamic neurodegeneration and promoted autophagy activation. Auotophagic inhibitor 3-methyladenine partially alleviated thalamic damage induced by SARM1. Moreover, autophagic initiation through rapamycin treatment aggravated post-stroke neuronal death and gliosis in Sarm1−/− mice. Taken together, SARM1 contributes to secondary thalamic neurodegeneration after cerebral infarction, at least partly through autophagy inhibition. SARM1 deficiency is a potential therapeutic strategy for secondary thalamic neurodegeneration and functional deficits after stroke.
Introduction
Ischemic stroke can cause acute ischemic damage as well as secondary neurodegeneration in remote regions with synaptic connections to the primary infarction. 1 For example, middle cerebral artery infarction causes neuronal damage in the ipsilateral thalamus outside the territory of the middle cerebral artery. 2 This secondary pathology may result from retrograde degeneration of the thalamocortical fibers rather than insufficient blood supply; however, the underlying molecular mechanisms remain unclear. Increasing evidence suggests that inflammatory reactions, oxidative stress, apoptosis, and excessive autophagy are involved in thalamic degeneration.3 –6 Notably, secondary neurodegeneration occurs several days or weeks after stroke onset and impedes the recovery of neurological function. 7 The wider tissue window for such lesions suggests that secondary neurodegeneration may be a promising therapeutic target for ischemic stroke.
Autophagy is a conserved lysosomal pathway that recycles damaged organelles and misfolded proteins to maintain energy metabolism and cell homeostasis. 8 We previously reported Beclin-1 and LC3-II upregulation as well as autophagosome accumulation in the ipsilateral thalamus at 7 and 14 days after middle cerebral artery occlusion (MCAO) in rats, which indicated autophagy activation. Furthermore, appliance of autophagic inhibitor 3-MA and Beclin-1 knockdown significantly reduced the post-MCAO thalamic neuronal loss and gliosis. 9 These findings suggest that autophagy overactivation is crucially involved in thalamic degeneration after ischemic stroke. However, the mechanisms underlying the excessive autophagy activation remain unclear.
The sterile α/Armadillo/Toll-Interleukin receptor homology domain protein (SARM1), a nicotinamide adenine dinucleotide (NAD+) hydrolase, is crucially involved in Wallerian degeneration. 10 Loss of SARM1 can suppress axonal degeneration and maintain long-term axonal survival both in vivo and in vitro. 11 Nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) deficiency resulting from transport hindrance after axotomy impairs the conversion of nicotinamide mononucleotide (NMN) into NAD+. 12 The increased NMN/NAD+ ratio finally activates SARM1 to trigger axonal degeneration. 13 Indeed, loss of SARM1 protects against axon degeneration in various models of neurological diseases, including amyotrophic lateral sclerosis (ALS), traumatic brain injury, and paclitaxel-induced peripheral neuropathy.14 –16 In the TDP-43Q331K transgenic mouse model of ALS-FTD, SARM1 deletion protects motor neuron cell bodies more than motor axons, which suggests that SARM1 may promote neuronal survival by maintaining retrograde trophic support. 14 Moreover, SARM1 is essential for the blockade of vesicle transport through damaged axons after nerve injury. dSarm null clones maintain the normal rates of axonal transport of autophagosomes from severed distal axons to cell bodies for ≥6 h after axotomy independent of NAD+ hydrolase activity. 17 However, it remains unclear whether SARM1 is involved in secondary thalamic degeneration and affects the autophagic-lysosomal pathway in the thalamus after focal cerebral infarction.
Herein, we aimed to investigate whether SARM1 loss could exert neuroprotective effects against thalamic neurodegeneration after focal cerebral infarction. Additionally, we aimed to examine the effects of SARM1 deletion on the autophagic-lysosomal pathway in the thalamus.
Methods
Animals
Sarm1−/− mice (B6.129X1-Sarm1tm1Aidi/J, strain:018069) mice were obtained from Jackson Laboratories and C57BL/6J wild-type mice were purchased from the Animal Center of Sun Yat-sen University. All animals were sheltered in a barrier system and kept on a 12 hours light/dark cycle at a constant temperature with access to water and food. All experimental protocols were approved by the Institutional Animal Ethical Committee of Sun Yat-sen University (SYSU-IACUC-2022-001556), following the Chinese Council on Animal Care guidelines and the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. Sarm1+/− mice were generated by crossing C57BL/6J wild-type mice with Sarm1−/− mice and bred to obtain age matched male Sarm1−/− (n = 38) and Sarm1+/+ (n = 39) littermate mice. While Sarm1−/− mice were also bred to obtain for the rest of male Sarm1−/− mice (n = 110). For all experiments, 8-12 weeks old male mice were used. Finally, photothrombotic infarction (n = 167) or sham (n = 20) procedures were performed on 187 male mice. 18
Surgical procedures
All mice underwent surgical procedures under anesthesia with 3.5% isoflurane for anesthesia induction and 1.0%–2.0% isoflurane for anesthesia maintenance. Body temperature was maintained at 37.0 ± 0.5 °C during the surgery and recovery periods using a heating pad. Regarding the photothrombotic procedure, the mice were injected with 0.2 mL of 10 mg/mL rose bengal (Sigma–Aldrich) into the tail vein, followed by 15 min of illumination using a cold light source with a 4-mm aperture placed 2 mm lateral to the bregma (0.0 mm). Mice in the sham group underwent a similar procedure, except that rose bengal was replaced with 0.2 mL of 0.9% saline. Five wild-type mice and four Sarm1−/− mice were excluded due to phtothrombotic failure (n = 7) or death within 7 days after surgery (n = 2).
Intrathalamic virus injection
Adeno-associated virus (AAV) encoding mouse Sarm1 with the hSyn promoter and negative control AAV-eGFP were constructed by Genechem (Shanghai, China). hSyn promoter specifically drives Sarm1 expression in neurons. AAV-Sarm1 or AAV-eGFP was stereotactically injected into the right thalamus of Sarm1−/− mice. Briefly, the mice were anesthetized as aforementioned and placed on a stereotactic apparatus. We used a 2.5 μL microsyringe (Hamilton, Cinnaminson, NJ) to inject the virus (2 μL; 1E + 12v.g./mL) into the thalamus at the following coordinates (four sites, 0.5 μL per site): 1.7 mm anteroposterior, 1.6 mm lateral, 3.3/3.7 mm dorsoventral; 2 mm anteroposterior, 1.7 mm lateral, and 3.3/3.7 mm dorsoventral relative to bregma. To allow sufficient gene expression, all injections were completed 1 month before photothrombotic infarction.
Pharmacological interference of autophagy
Rapamycin, which is an autophagic inducer, inhibits phosphorylation of the mammalian target of rapamycin (mTOR) to enhance autophagic flow activation. 19 3-methyladenine (3-MA), an autophagic inhibitor, selectively inhibits the activation of PI3K to block autophagic flow activation. 20 Rapamycin (Sigma–Aldrich, V900930-1MG) or 3-MA (Sigma-Aldrich, M9281) was dissolved in 2% DMSO.21,22 Rapamycin (20 μg/2 μL), 3-MA(25 nmol/5 μL) or the respective solvents were slowly injected into the right ventricle of Sarm1−/− mice using a 10-μl syringe with an infusion rate of 0.4 μL/min at 24 h after photothrombotic infarction. After infusion, the syringe was left in place for 5 minutes to allow drug diffusion.
Neurological evaluation
Neurological function was evaluated using modified neurological severity scores (mNSS) and the adhesive-removal test at days 1, 3, 7, and 14 after photothrombotic infarction (n = 6 per group at each time point). The mNSS comprised motor, sensory, reflex, and beam balance tests and was graded on a scale of 0 to 18 (from no deficit [0] to maximal deficit [18]). 23 The adhesive-removal test was used to assess the sensorimotor function of each forepaw. 24 Briefly, two stickers were attached to each forelimb and the removal time was recorded. Before photothrombotic surgery, all the mice were trained until they could remove the stick within 10 s.
Infarct volume
To evaluate infarct volume, in vivo MRI was performed on the Aspect M3 system at 3, 7, and 14 days after photothrombotic infarction (Aspect Imaging, Israel). Images were collected using a fast spin-echo T2-weighted sequence with the following parameters: echo time/repetition time = 76.67/ 7663 ms and 15 slices with 1-mm slice thickness. Infarct volume was calculated using Image J software with correction for brain swelling using the equation: [(area of the contralateral hemisphere – area of the ipsilateral hemisphere)/area of the contralateral hemisphere] × 100%. The lesions were summed and multiplied by the slice thickness to determine the infarct volume.
Immunohistochemistry and Nissl staining
Mice placed under deep anesthesia were transcardially perfused with 0.9% saline at 4 °C followed by 4% paraformaldehyde. After post-fixing for 8 h at 4 °C, the brains were sequentially immersed in 20% and 30% sucrose until they sunk. Brains slices (10-μm thick) were cut on a cryostat (CM1900, Leica, Heidelberger, Germany) and prepared for immunostaining and Nissl staining.
The slides were incubated with bovine serum albumin containing 0.3% Triton X-100 for 1 h to block non-specific binding. Subsequently, brain sections were incubated overnight at 4 °C with the following rabbit and mouse primary antibodies: rabbit anti-NeuN (neuronal marker, 1:500, Cell Signaling Technology, 24307), chicken anti-GFAP (astrocyte marker, 1:1500, Abcam, ab4674), mouse anti-Iba-1 (microglia marker, 1:500,Wako, 019-19741), goat anti-Iba-1 (microglia marker, 1:500,abcam, ab5076), mouse anti-NeuN (1:500, Abcam, ab104224), rabbit anti-SARM1 (1:500, Cell Signaling Technology, 13022), LC3B (1:200, Cell Signaling Technology, 83506), and LAMP1 (1:200, Abcam, ab24170). Next, the sections were rinsed in 0.01 M phosphate-buffered saline (PBS), followed by incubation with the following secondary antibodies at 24–26 °C for 60 min: Alexa Fluor 594-conjugated goat anti-mouse IgG (1:500, Abcam, ab150116), Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:500; Abcam, ab150077), Alexa Fluor 555-conjugated goat anti-rabbit IgG (1:500; Abcam, ab150078), Alexa Fluor 674-conjugated goat anti-chicken IgG (1:500; Abcam, ab150175), Alexa Fluor 674-conjugated donkey anti-goat IgG (1:500; Abcam, ab150131), and Alexa Fluor 555-conjugated donkey anti-rabbit IgG (1:500; Abcam, ab150062). Nissl staining was performed using 0.3% cresyl violet (Beyotime, China) in a series of sections from bregma −1.3 to −1.9 mm to assess neuronal damage in the ipsilateral thalamus.
Transmission electron microscopy
Mice under deep anesthesia were sequentially perfused with 2.5% ice-cold glutaraldehyde in 0.1 M PBS. The right thalamus was quickly separated and cut into 1-mm³ blocks. After fixing in 1% osmium tetroxide, samples were dehydrated in graded ethyl alcohol and embedded in epoxy resin. Finally, ultra-thin (80 nm) cross-sections were stained using saturated uranyl acetate and lead citrate, followed by scanning using a transmission electron microscope (FEI, Tecnai G2 Spirit Twin).
Western blot
Four mice from each group were sacrificed at 7 or 14 days after photothrombotic surgery. All mice underwent intracardiac perfusion with 0.9% saline, followed by separation of the right thalamus on ice. Total protein was extracted from the thalamus using radioimmunoprecipitation assay buffer (Beyotime, China) with 1 mM protease inhibitor (Boster, China). Protein concentrations were determined using the BCA Protein Assay Kit (Thermo Fisher, USA). We extracted 30 μg of protein from each sample, separated it using SDS-PAGE, and transferred it onto a polyvinylidene fluoride membrane (Millipore, ISEQ00010). Subsequently, membranes were blocked with 5% non-fat dried milk at room temperature for 1 h, followed by incubation using the following primary antibodies: rabbit anti-SARM1 (1:500, Cell Signaling Technology, 13022), rabbit anti-NMNAT2 (1:1000, Abcam, ab56980), rabbit anti-LC3 (1:1000, Abcam, ab192890), mouse anti-P62/SQTSM1 (1:1000, Abcam, ab56416), rabbit anti-beclin 1 (1:1000, Proteintech, 11306-1-AP), rabbit anti-LAMP1(1:1000, Abcam, ab24170), rabbit anti-cathepsin D (1:1000, Abcam, ab75852), rabbit anti-Atg14 (N-terminal, 1:1000, Protein tech, 19491-1-AP), rabbit anti-rab7 (1:1000, Protein tech, 55469-1-AP), rabbit anti-GAPDH (1:1000; Cell Signaling Technology, 2118S), or rabbit anti-TUBA/ɑ-tubulin (1:1000; Cell Signaling Technology, 2125S) overnight at 4 °C. Next, the membranes were incubated using the corresponding secondary antibodies for 60 min at 24–26 °C: goat anti-mouse IgG (1:2000; Cell Signaling Technology, ab6789) or goat anti-rabbit IgG (1:2000; Cell Signaling Technology, 7074). Finally, membranes were washed using Tris-buffered saline with 0.1% Tween® 20 Detergent; moreover, immunoreactivity was detected using an enhanced chemiluminescence system (Millipore, WBKLS0100).
Quantification
Fiji (National Institutes of Health, Bethesda, MD, USA) software was used to analyze the Nissl staining, immunostaining, and immunoblotting results. The number of intact neurons and NeuN+ cells in the ipsilateral thalamus was counted within three non-overlapping fields (294 × 294 μm) under ×400 magnification and presented as the average cell number per field in each section. The immunoreactivity of GFAP+ and Iba-1+ cells was calculated within three non-overlapping fields (294 × 294 μm) under ×400 magnification and presented as the percentage of GFAP-immunopositive and Iba-1-immunopositive areas per field in each section.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0 (GraphPad, San Diego, CA, USA). Continuous data are expressed as mean ± standard deviation (SD). The normality of each dataset was assessed by Shapiro-Wilk test. Between-group differences were analyzed using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test, or two-way ANOVA with Bonferroni's post-hoc test for multiple comparisons. Statistical significance was set at a two-sided P < 0.05.
Results
Functional recovery is improved in Sarm1−/− mice after photothrombotic infarction
First, we investigated whether Sarm1−/− mice showed ameliorated neurological deficits after photothrombotic infarction. The degree of neurological deficits was evaluated using the mNSS and adhesive-removal tests at baseline as well as 1, 3, 7, and 14 days after photothrombosis (Figure 1(a)). There was no difference in the baseline neurological scores between Sarm1−/− and WT mice. At 1 post-stroke day, both groups developed neurological deficits, which were indicated by increased mNSS scores and latency to remove the tape; however, there was no significant between-group difference (Figure 1(b) and (c)). However, there was a between-group distinction in the temporal evolution of neurological deficits. Compared with WT mice, Sarm1−/− mice showed a significant decrease in the mNSS score and tape removal time at 3, 7, and 14 days after stroke (Figure 1(b) and (c)). These results indicated that Sarm1−/− mice showed better neurological recovery after photothrombotic infarction. To investigate whether the difference in functional recovery was related to infarct size, we performed in vivo MRI scans at 3, 7, and 14 postoperative days. Cerebral infarction was detected as a hyperintense area on T2-weighted images and was consistently restricted to the right cortex (Figure 1(d)). The photothrombosis-induced infarct volume decreased from day 3 to 14 after ischemia in both Sarm1−/− and WT mice (Figure 1(e)). No significant between-group difference was observed in infarct volume, which suggested that the better functional recovery in Sarm1−/− mice may not be related to the primary infarction (Figure 1(e)).
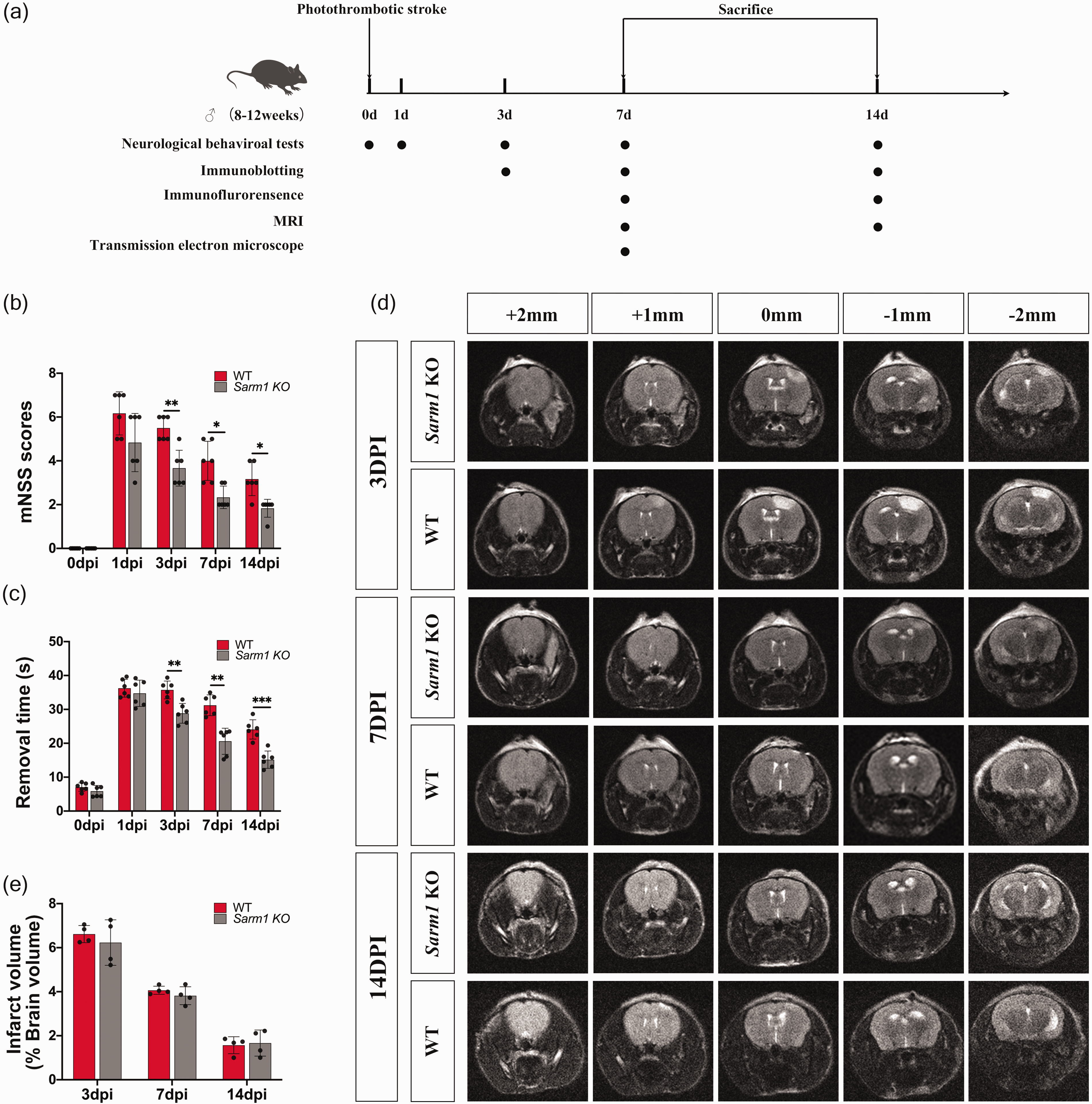
Sarm1−/− mice showed improved functional recovery after photothrombotic infarction. (a) Schematic diagram of the experimental design. (b-c) Neurological deficits were measured using the modified Neurological Severity Scores (mNSS) and adhesive-removal test in Sarm1−/− and wild-type (WT) mice at 3, 7, and 14 days after photothrombotic infarction. N = 6 mice for each time point per group. (d) Representative brain MR images from bregma −2 mm to + 2 mm indicated cerebral cortical infarction in the right hemisphere and (e) Quantitative analysis of infarct volume in Sarm1−/− and WT mice at 3, 7, and 14 days after photothrombotic infarction. N = 4 mice for each time point per group. Data are shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, compared with the WT group. Two-way ANOVA with Bonferroni’s post-hoc test.
Thalamic neuronal death and gliosis are attenuated in Sarm1−/− mice after cerebral infarction
To investigate the effect of Sarm1 deletion on post-infarction secondary thalamic damage, we determined the SARM1 expression pattern in the ipsilateral thalamus. Double immunostaining showed that SARM1 was predominantly expressed in neuron-specific NeuN+ cells and partly expressed in astrocyte-specific GFAP+ cells in the ipsilateral thalamus of WT mice at 7 post-stroke days (Figure 2(a)). Contrastingly, there was rare colocalization of SARM1 with microglia-specific Iba1+ cells (Figure 2(a)). Moreover, immunoblotting did not detect SARM1 protein in the ipsilateral thalamus of Sarm1−/− mice, which demonstrated successful knockout of Sarm1 (Figure 2(b) and (c)). Compared with the sham-operated animals, WT mice did not show altered thalamic SARM1 levels at 7 post-stroke days; however, they started to decrease at 14 post-stroke days (Figure 2(b) and (c)). Additionally, there was a substantial post-stroke decrease in NMNAT2, which is an axonal survival factor, in both Sarm1−/− and WT mice (Figure 2(d)).
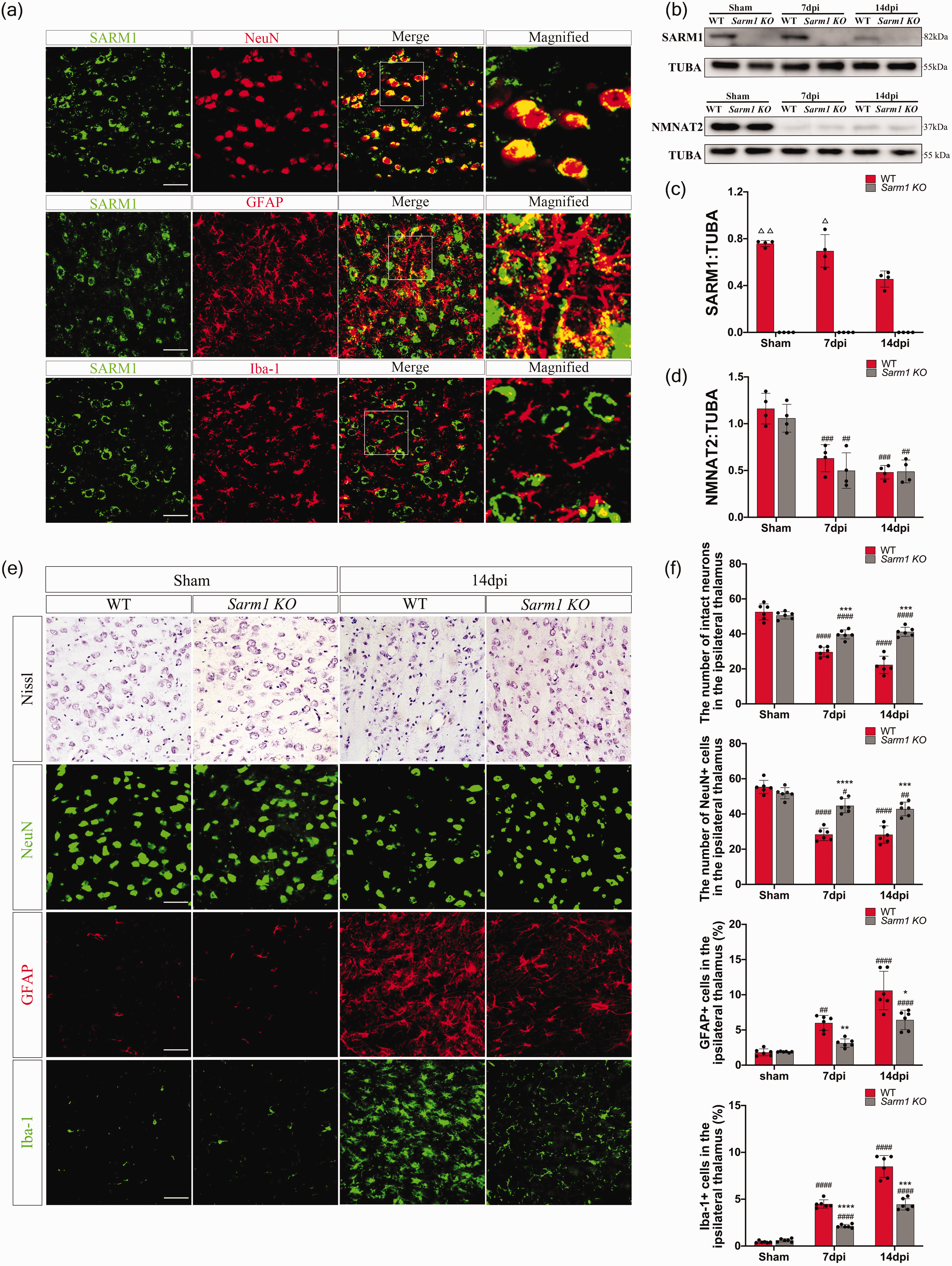
Thalamic neuronal death and gliosis are attenuated in Sarm1−/− mice after cerebral infarction. (a) Double immunostaining for SARM1 and cell-specific markers in the ipsilateral thalamus at 7 days after cerebral infarction. SARM1 was mainly expressed by NeuN+ neurons and partly by GFAP+ astrocytes in the ipsilateral thalamus at 7 post-stroke days. SARM1 was not colocalized with Iba1+ microglia. (b) Western blot showed SARM1 and NMNAT2 expression in the ipsilateral thalamus of Sarm1−/− and WT mice Continued.after photothrombotic infarction or sham procedure. (c) Quantitative analysis of SARM1 protein levels relative to a-tubulin. N = 4 mice for each time point per group. (d) Quantitative analysis of the NMNAT2 protein level relative to TUBA. N = 4 mice for each time point per group. (e) Representative images of Nissl, NeuN, GFAP, and Iba-1 staining in the ipsilateral thalamus of WT and Sarm1−/− mice and (f) Quantification of intact neurons, NeuN+ cells, GFAP+ cells, and Iba-1+ cells in the thalamus. N = 6 mice for each time point per group. Data are shown as mean ± SD, ▵P < 0.05, ▵▵P < 0.01, compared with WT group at 14 days post stroke, one-way ANOVA with Tukey’s multiple comparisons test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, compared with WT group; two-way ANOVA with Bonferroni’s post-hoc test. ##P < 0.01, ###P < 0.001, ####P < 0.0001; compared with respective sham group; one-way ANOVA with Tukey’s multiple comparisons test. Scale bar = 50 μm.
Next, we explored changes in secondary thalamic degeneration after Sarm1 deletion. Nissl staining revealed that some cells showed shrunken cell bodies and condensed nuclei in the ipsilateral thalamus of WT mice at 7 and 14 post-stroke days. Compared with WT mice, Sarm1−/− mice showed an increased number of intact neurons in the ipsilateral thalamus at both time points (Figure 2(e) and (f)). Similarly, Sarm1−/− mice showed a higher number of thalamic NeuN+ cells than WT mice (Figures 2(e) and (f)). Moreover, WT mice showed remarkable post-stroke activation of reactive astrocytes and microglia in the ipsilateral thalamus (Figures 2(e) and (f)). Compared with WT mice, Sarm1−/− mice showed a significantly reduced number of thalamic astrocytes and microglia at 7 and 14 post-stroke days (Figures 2(e) and (f)). Taken together, the loss of SARM1 is associated with reduced secondary thalamic degeneration after stroke.
Autophagy activation after cerebral infarction is inhibited in the ipsilateral thalamus of Sarm1−/− mice
Given the involvement of autophagy in thalamic degeneration, we investigated whether the loss of SARM1 affects the autophagic-lysosomal pathway in the thalamus after cerebral infarction. Consistent with previous findings, WT mice showed significantly increased LC3B-II and Beclin1 protein levels in the ipsilateral thalamus at 7 post-stroke days, which returned to baseline levels at 14 days (Figure 3(a) and (b)). Compared with WT mice, Sarm1−/− mice showed significantly decreased LC3-II and Beclin1 levels in the ipsilateral thalamus, indicating an association between the loss of SARM1 and suppression of autophagic activation (Figure 3(a) and (b)). Transmission electron microscopy revealed axonal and cellular swelling in the ipsilateral thalamus of WT mice after cerebral infarction. Further, WT mice showed accumulation of numerous double-membrane autophagosomes in the swollen axons and soma of thalamic neurons ipsilateral to the infarction side, which were few in Sarm1−/− mice or sham group mice (Figure 3(c)). Additionally, we examined the expression of the autophagosome substrate SQSTM1/P62 as an autophagic flux marker. SQSTM1/P62 levels were substantially elevated at 7 post-stroke days; moreover, Sarm1−/− mice showed lower SQSTM1/P62 levels in the ipsilateral thalamus than WT mice (Figure 3(a) and (b)).
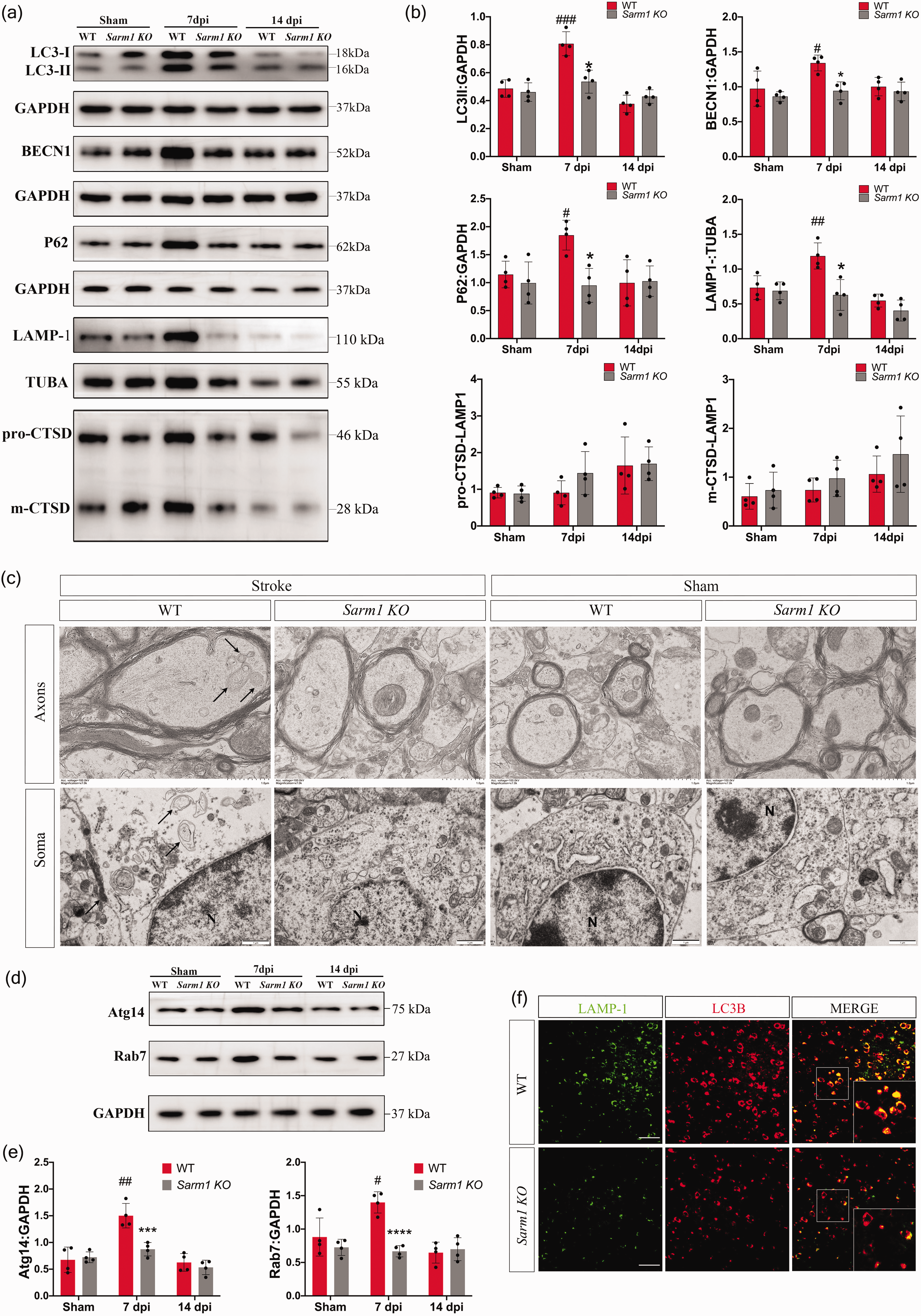
Autophagy activation is inhibited in the thalamus of Sarm1−/− mice after cerebral infarction. (a) Western blot showing protein expression of LC3, P62, Beclin1, LAMP-1, and cathepsin D (CTSD) in the ipsilateral thalamus in the sham control and ischemic Continued.Sarm1−/− and WT mice. (b) Quantitative analysis of LC3 II, P62, and Beclin1 protein levels relative to GAPDH; LAMP-1 protein levels relative to a-tubulin; and pro-CTSD and m-CTSD levels relative to LAMP-1 levels. N = 4 mice for each time point per group. (c) Representative electron microscopy images for neuronal axons and somas in the ipsilateral thalamus of Sarm1−/− and WT mice. The black arrows indicate autophagosomes. Scale bar = 1 μm. (d) Western blot showing protein expression of Atg14 and Rab7 in the ipsilateral thalamus in the sham control and ischemic Sarm1−/− and WT mice. (e) Quantitative analysis of Atg14 and Rab7 protein levels relative to GAPDH; N = 4 mice for each time point per group and (f) Colocalization of LC3B with LAMP-1 in the ipsilateral thalamus at 7 post-infarction days. Data are shown as mean ± SD, *P < 0.05, ***P < 0.001, ****P < 0.0001, compared with the WT group, two-way ANOVA with Bonferroni’s post-hoc test. Scale bar = 50 μm. Data are shown as mean ± SD, #P < 0.05, ##P < 0.01, ###P < 0.001, compared with respective sham group; one-way ANOVA with Tukey’s multiple comparisons test.
Autophagosomes fuse with lysosomes to facilitate the degradation of proteins and organelles. To investigate the role of lysosomes in autophagosome accumulation, we examined the expression of LAMP-1 and cathepsin D (CTSD), which are markers of lysosomes and lysosomal proteases, respectively. CTSD, a soluble lysosomal aspartic endopeptidase, is crucially involved in lysosomal clearance and degradation through the conversion of the cathepsin D precursor (pro-CTSD) to mature cathepsin D (m-CTSD) in lysosomes. 25 At 7 post-stroke days, WT mice showed upregulated LAMP-1 protein levels in the ipsilateral thalamus, which returned to baseline levels at 14 post-stroke days (Figure 3(a) and (b)). Furthermore, there was no post-stroke increase in thalamic pro-CTSD/LAMP1 and m-CTSD/LAMP1 levels, indicating no alteration in the lysosomal enzyme function. Loss of SARM1 prevented the transient elevation of LAMP-1 protein and did not affect pro-CTSD/LAMP1 and m-CTSD/LAMP1 levels (Figure 3(a) and (b)). Accessory protein Atg14 and Rab7 is essential for the fusion of autophagasomes and lysosomes. WT mice showed increased Atg14 and Rab7 protein levels in the ipsilateral thalamus along with autophagic activation at 7 post-stroke days, which returned to baseline levels at 14 post-stroke days (Figure 3(d) and (e)). Immunofluorescence staining further demonstrated post-stroke colocalization of LAMP-1 with most LC3B+ cells in WT mice. Moreover, Sarm1−/− mice showed reduced double immunostaining of LAMP-1 and LC3B cells in the ipsilateral thalamus (Figure 3(f)). Taken together, these findings implied that autophagosome-lysosome fusion in the thalamus was not impaired after stroke.
AAV-Sarm1 injection induces neuronal death, gliosis, and autophagy activation in Sarm1−/− mice
To further examine the effect of SARM1 on secondary thalamic neurodegeneration, we performed gene-rescue experiments by restoring thalamic SARM1 expression of Sarm1−/− mice at 1 month before photothrombotic surgery (Figure 4(a)). SARM1 was successfully re-expressed in the ipsilateral thalamus at 7 and 14 post-stroke days (Figure 4(b)). Immunostaining revealed colocalization of eGFP-SARM1 with NeuN+ cells in the thalamus but no expression of eGFP-SARM1 in astrocytes or microglia, which indicated selective SARM1 expression in neurons (Figure 4(c)).
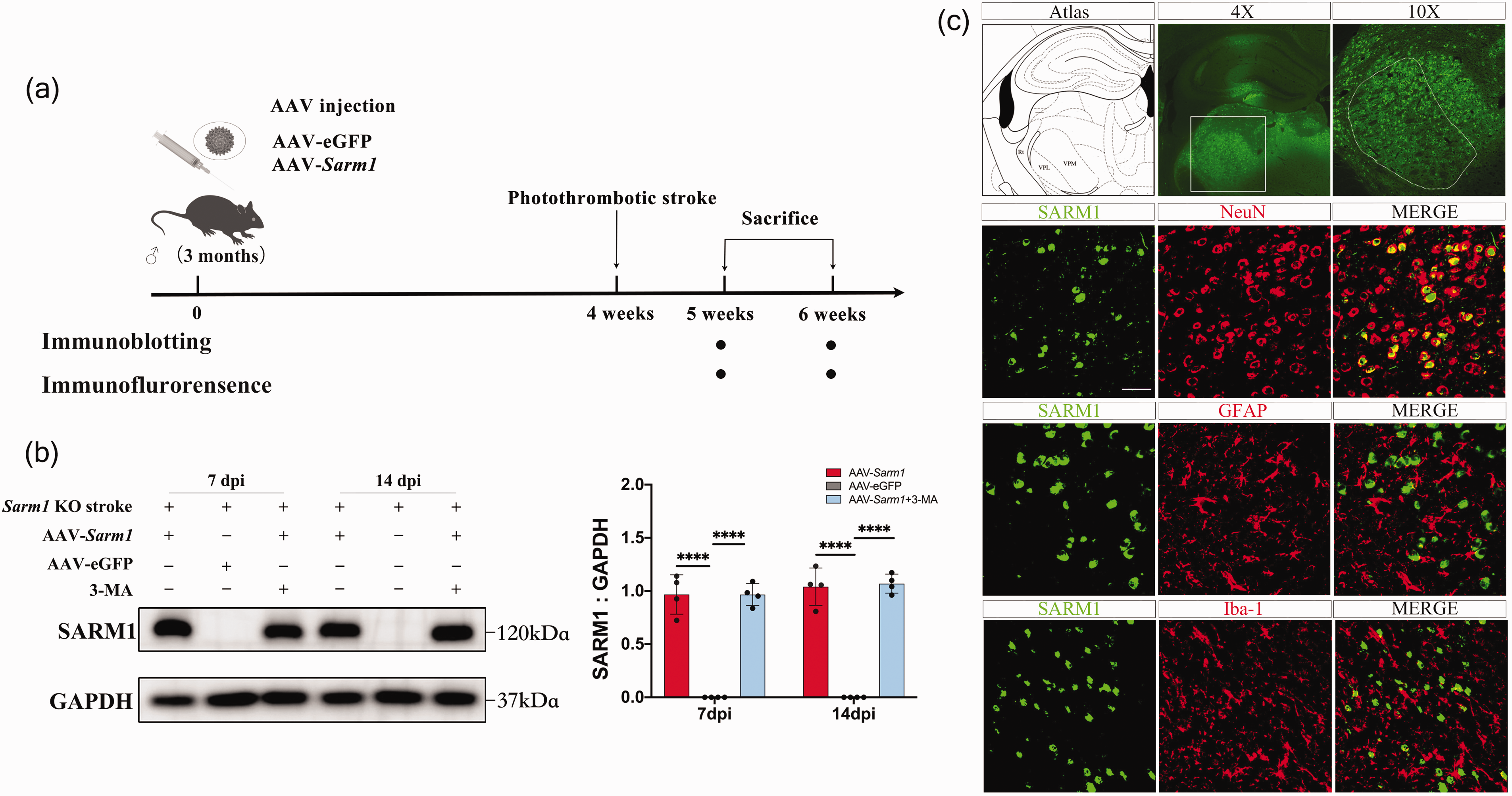
Successful SARM1 re-expression in thalamic neurons of AAV-Sarm1 group. (a) Schematic diagram of the experimental design. AAV-eGFP and AAV-Sarm1 were injected into the thalamus ipsilateral to the infarction in Sarm1−/− mice. (b) Western blot showed SARM1 expression in the ipsilateral thalamus of AAV-eGFP, AAV-Sarm1 and AAV-Sarm1 + 3-MA groups at 7 and 14 post-infarction days and (c) EGFP-tagged SARM1 were detected in the ipsilateral thalamus at 7 days after MCAO; moreover, EGFP-tagged SARM1 were colocalized with NeuN+ cells but not with GFAP+ or Iba-1+ cells. Scale bar: 50 μm. N = 4 mice for each time point per group. Data are shown as mean ± SD, ****P < 0.0001, two-way ANOVA with Bonferroni’s post-hoc test. VPL, ventroposterior thalamic nuclei, lateral; VPM, ventroposterior thalamic nuclei, medial.
The results of behavioral tests showed the re-expression of SARM1 caused a siginificant increase in the mNSS score and tape removal time at 3, 7, and 14 days after stroke (Figure 5(a)). Furthermore, compared with AAV-Sarm1 group, additional administration of 3-MA siginificantly decreased the tape removal time since 3 days post stroke. These results suggest that SARM1 re-expression enhanced the neurological deficits and autophagic inhibitor 3-MA partially rescued the somatosensory deficits of mice after photothrombotic stroke. Compared with control AAV, SARM1 re-expression strikingly reduced the number of thalamic NeuN+ cells at 7 and 14 post-stroke days (Figure 5(b) and (c)). Additionally, thalamic GFAP+ and Iba-1+ cells were markedly increased at all time points (Figure 5(b) and (c)). While 3-MA treatment increases thalamic neuronal survivals and reduced gliosis after AAV-Sarm1 injection.
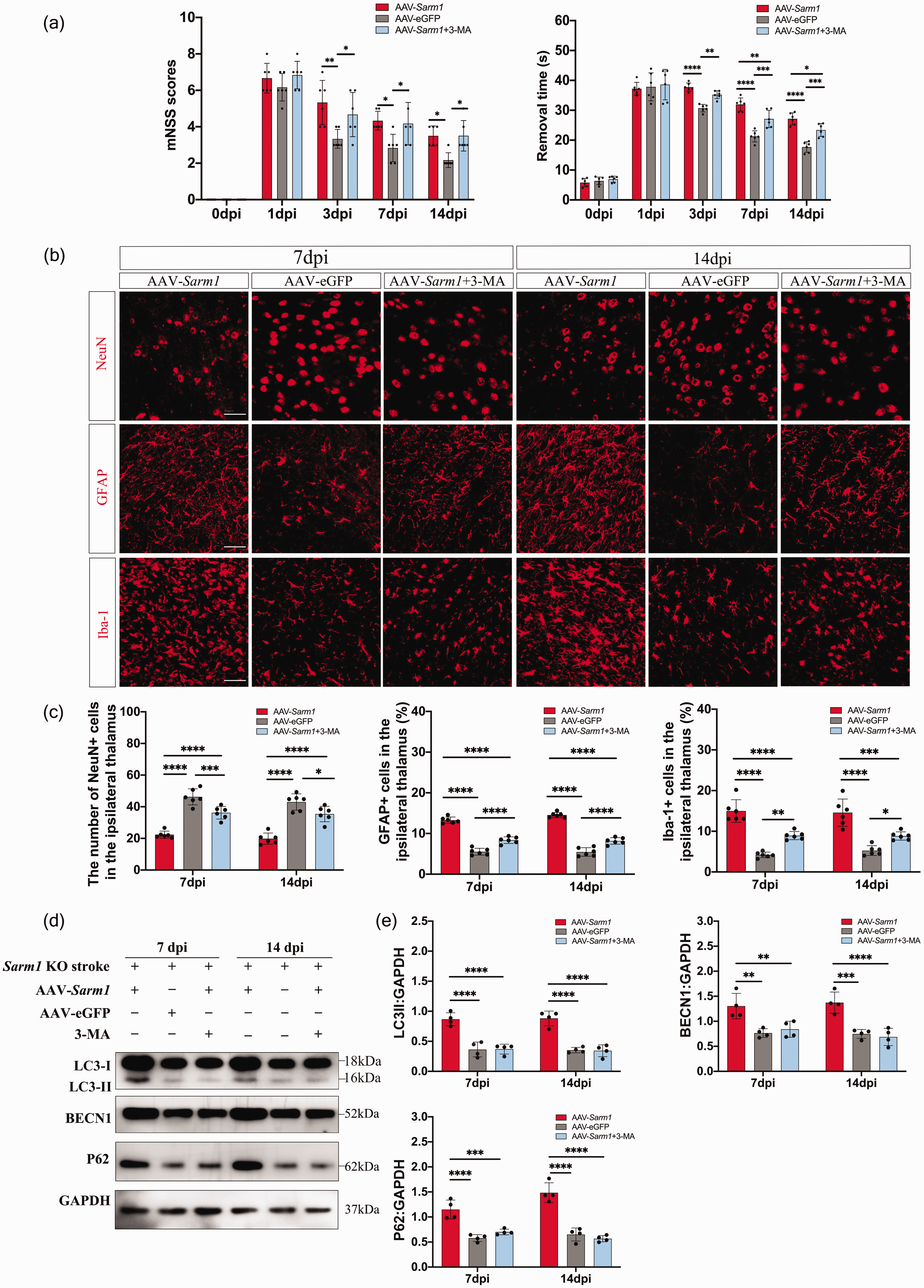
AAV-Sarm1 injection induced neuronal death, gliosis, and autophagy activation in Sarm1−/− mice. (a) Neurological deficits were measured using the modified Neurological Severity Scores (mNSS) and adhesive-removal test in AAV-eGFP, AAV-Sarm1 and Continued.AAV-Sarm1 + 3-MA groups at 3, 7 and 14 days after photothrombotic infarction. N = 6 mice for each time point per group. (b) Representative images of NeuN, GFAP, and Iba-1 immunostaining in the ipsilateral thalamus of AAV-eGFP, AAV-Sarm1 and AAV-Sarm1+ 3-MA groups (scale bar, 50 μm). (c) Quantification of NeuN+, GFAP+, and Iba-1+ cells in the ipsilateral thalamus. N = 6 mice for each time point per group. (d) Western blot analysis showed LC3, P62, and Beclin1 expression in the ipsilateral thalamus of AAV-eGFP, AAV-Sarm1 and AAV-Sarm1 + 3-MA groups at 7 and 14 post-stroke days and (e) Quantitative analysis of SARM1, LC3 II, Beclin1, and P62 levels relative to GAPDH levels. N = 4 mice for each time point per group. Data are shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two-way ANOVA with Bonferroni’s post-hoc test.
Next, we investigated the effect of SARM1 re-expression on autophagy. At 7 and 14 post-stroke days, LC3B-II, Beclin-1, and SQSTM1/P62 protein levels were significantly increased in the ipsilateral thalamus after AAV-Sarm1 injection compared with AAV-eGFP injection, while 3-MA administration downregulated these proteins to the level of AAV-eGFP group. (Figure 5(d) and (e)). Taken together, these findings supported the involvement of SARM1 in secondary damage and excessive autophagy in the thalamus following cerebral infarction. The reoccurrence of thalamic neurodegeneration after neuron-specific expression of SARM1 suggests cell-autonomous SARM1 activity within neurons mediates secondary thalamic neurodegeneration after stroke, which can be partially rescued by inhibition of autophagy via 3-MA adiministration.
Treatment with rapamycin attenuates the protective effect of Sarm1 deletion on post-stroke thalamic neurodegeneration
To investigate whether excessive autophagy activation acts downstream of SARM1 to induce secondary thalamic neurodegeneration, rapamycin was administered at 24 hours post-stroke in Sarm1−/− mice. The administration of rapamycin caused a siginificant increase of mNSS score and tape removal time at 3 and 7 days after stroke. In contrast, rapamycin treatment did not lead to neurological deficits in mice subjected to sham procedure (Figure 6(a)). Rapamycin remarkably increased LC3B-II conversions in the ipsilateral thalamus at 7 days after photothrombotic or sham procedure, which indicates autophagy activation (Figure 6(b) and (c)). Additionally, rapamycin treatment decreased the number of NeuN+ cells and increased GFAP+ as well as Iba-1+ cells in the ipsilateral thalamus of Sarm1−/− mice at 7 days after stroke (Figure 6(d) and (e)). In contrast, rapamycin did not increase the neuronal loss and gliosis in the thalamus of sham-operated mice. These results indicated rapamycin enhanced the thalamic neurodegeneration after stroke, possibly due to increased autophagy activation downstream of stroke-induced SARM1 activity rather than direct neuronal toxicity of rapamycin.
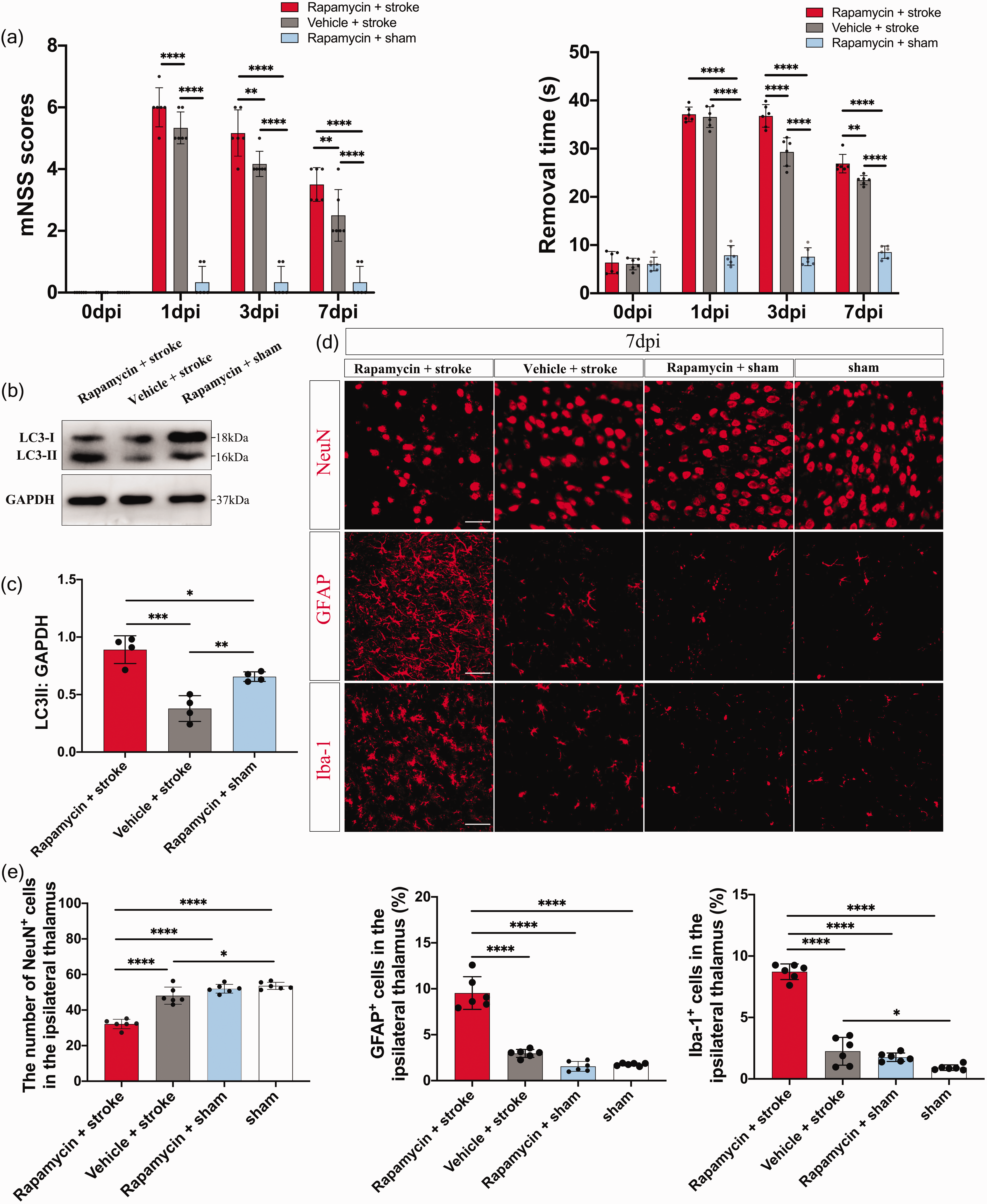
Treatment with rapamycin attenuates the protective effect of Sarm1 deletion on post-stroke thalamic neurodegeneration. (a) Neurological deficits were measured using the modified Neurological Severity Scores (mNSS) and adhesive-removal test at 3, 7 and 14 days after photothrombotic infarction. N = 6 mice for each time point per group. (b) Western blot showing LC3 expression in the ipsilateral thalamus of rapamycin + stroke, vehicle + stroke and rapamycin + sham groups at 7 post-stroke days. (c) Quantitative analysis of LC3 II level relative to GAPDH levels. N = 4 mice per group. (d) Representative images of NeuN, GFAP, and Iba-1 immunostaining in the ipsilateral thalamus of rapamycin + stroke, vehicle + stroke, rapamycin + sham and sham groups. Scale bar = 50 μm and (e) Quantification of NeuN+, GFAP+, and Iba-1+ cells in the ipsilateral thalamus. N = 6 mice per group. Data are shown as mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; two-way ANOVA with Bonferroni’s post-hoc test; one-way ANOVA with Tukey’s multiple comparisons test.
Discussion
We found that Sarm1 gene deletion alleviated neurological deficits induced by photothrombotic cerebral cortical infarction. Although no reduction was observed in the infarct volume, secondary thalamic neurodegeneration was substantially ameliorated by Sarm1 gene deletion. The protective effect of Sarm1 deficiency on thalamic neurodegeneration vanished after thalamic SARM1 re-expression. Moreover, loss of SARM1 is related to the suppression of excessive autophagic activation. The inhibition of autophagy reduced SARM1-mediated thalamic neurodegeneration, and induction of autophagic activation attenuated the protective effect of SARM1 deficiency on thalamic neurodegeneration following photothrombotic infarction. Taken together, our findings demonstrate that SARM1 inactivation is a potential therapeutic strategy for secondary thalamic neurodegeneration and post-stroke functional deficits.
Currently, the therapeutic strategy for acute ischemic stroke is rescuing the ischemic penumbra around the infarcted core through immediate recanalization of the occluded arteries using thrombolytic agents or endovascular thrombectomy. 26 However, most patients with acute ischemic stroke are ineligible for effective treatment since they have passed the therapeutic time window. 27 In addition to acute ischemic damage, ischemic stroke causes delayed neurodegeneration in non-ischemic regions that have synaptic connections to the primary infarction site, including the thalamus. Secondary neurodegeneration occurs several days to weeks after stroke; therefore, it has a relatively wide therapeutic time window. Unfortunately, there are few strategies targeting this secondary neurodegeneration following ischemic stroke. We found improved functional recovery and attenuated thalamic neurodegeneration in Sarm1−/− mice after photothrombotic infarction. This suggests that SARM1 may be a novel neuroprotective target for the treatment of acute ischemic stroke.
Inhibition of SARM1 phosphorylation through serine 548 alanine mutation was found to reduce the infarct size, neuronal death in the ischemic penumbra, and neurobehavioral dysfunction in rats after cerebral ischemia/reperfusion (I/R) injury. 28 However, in our study, Sarm1 gene deletion did not significantly affect the infarct volume after photothrombotic infarction. This inconsistency could be attributed to differences in the stroke model between the studies. A previous study established an MCAO/reperfusion model using a suture-occluded method, which has a large infarct size with cerebral edema, ischemic penumbra, and I/R injury. Unlike MCAO models, the photothrombotic stroke model has a narrow/absent salvageable ischemic penumbra given the rapid development of tissue necrosis and edema. 29 Photothrombotic surgery induces infarctions predominantly restricted to the cerebral cortex; accordingly, it is suitable for examining secondary neurodegeneration in remote regions. A transient MCAO model may be better for investigating whether the loss of Sarm1 rescues the ischemic penumbra or alleviates reperfusion injury. Furthermore, MRI did not detect signal abnormality within or surrounding the ipsilateral thalamus after photothrombotic infarction, which demonstrated that thalamic damage does not result from cerebral edema or blood insufficiency. Since infarct volume was not alleviated, the improved functional recovery in Sarm1−/− mice may be related to the attenuation of secondary thalamic degeneration.
SARM1, an NAD+ hydrolase, is the central executioner of pathological axonal degeneration.10,11 NMNAT2 is a crucial axonal survival factor that is rapidly degraded following various types of axonal injuries, including mechanical injury, chemotherapy, and mitochondrial toxins. 30 NMNAT2 loss reduces the NAD+/NMN ratio, which subsequently activates SARM1 for axonal self-destruction.13,31 SARM1 has an N-terminal region containing heat/armadillo (ARM) domains, two central sterile motifs (SAM), and a C-terminal toll/interleukin-1 receptor (TIR) domain. 32 Upon axonal injury, the auto-inhibitory of TIR by ARM domains are released.This causes TIR dimerization, which cleaves NAD+ to ADP-ribose (ADPR) and cyclic ADPR (cADPR). 32 In our study, thalamic NMNAT2 levels were substantially reduced in both Sarm1−/− and WT mice after cerebral infarction; however, thalamic neurodegeneration was ameliorated in Sarm1−/− mice. This suggests that SARM1 acts downstream of NMNAT2 to induce axonal degeneration. Moreover, thalamic SARM1 levels remained stable for 7 post-stroke days, followed by a reduction at 14 post-stroke days, which could be attributed to progressive neuronal death in the thalamus. Therefore, SARM1 activation may not require increased protein expression and may represent an early event in thalamic neuronal death.
Autophagy inhibition through 3-MA or Beclin1 knockdown attenuates thalamic degeneration following permanent MCAO in rats.3,9 Accordingly, we observed thalamic autophagy induction in WT mice after photothrombotic cerebral infarction. Although the increased SQSTM1/P62 levels suggested impaired autophagosome degradation, further examination of lysosomal function revealed an increased number of lysosomes and normal lysosomal proteinase levels. Atg14 and Rab7, which were directly involved in the autophagosome-lysosome fusion, increased in the ipsilateral thalamus at 7 post-stroke days and returned to baseline levels at 14 post-stroke days. Double immunostaining also revealed good colocalization of LAMP-1 with LC3 immunofluorescence in thalamic cells of WT mice. Therefore, photothrombotic cerebral infarction may not impair thalamic autophagosome clearance. The increased SQSTM1/P62 levels may be partly attributed to oxidative stress in thalamic cells. SQSTM1/P62 is a stress-inducible protein that can be upregulated via the Keap1-Nrf2 pathway for antioxidant response. 33 Oxidative damage contributes to thalamic degeneration following MCAO. 34 Therefore, SQSTM1/P62 upregulation may represent a response to oxidative stress rather than a disruptive autophagic flux.
How SARM1 mediates thalamic neurodegeneration following cerebral infarction remains unclear. The downstream degeneration executors could be loss of NAD+ as well as generation of ADPR, cADPR, or other molecules. 35 SARM1 deletion alleviated thalamic damage and was accompanied by autophagy inactivation. Moreover, thalamic SARM1 expression in Sarm1−/− mice reproduced thalamic degeneration and autophagy activation. Inhibition of autophagy by 3-MA administration after SARM1 expression partially rescued the thalamic degeneration and treatment with rapamycin aggravated thalamic degeneration in Sarm1−/− mice following cerebral infarction. Therefore, autophagy may act downstream of SARM1 and contribute to secondary thalamic damage. The mechanisms underlying the SARM1-induced modulation of autophagy remain unclear. Upon activation, SARM1 hydrolyzes NAD+ to generate ADPR and cADPR, which significantly reduces NAD+ levels. NAD+ is a potential regulator of ATP production. Energy deficits (i.e., a decrease in ATP levels or the AMP/ATP ratio) can rapidly activate AMPK and induce autophagy. 36 NAD+ supplementation can block autophagic activation and reduce ischemic damage. 37 Additionally, ADPR and cADPR are powerful calcium mobilizers that are crucially involved in paclitaxel-induced axon degeneration. 38 An increase in intracellular calcium levels may activate autophagy. 39 Therefore, NAD+ deficiency and ADPR or cADPR production may modulate the autophagic-lysosomal pathway. Further studies are warranted to elucidate the underlying molecular mechanisms.
There are some limitations in this study. Firstly, the sample size in each group is relatively small. Some groups include less than 6 mice, which could be underpowered for stroke studies. Secondly, only male mice are used in this study. It is unknown whether the loss of SARM1 is protective from secondary thalamic neurodegeneration in female ischemic stroke. Thirdly, memory tests are not included in the study. Thalamocortical communication has been verified as an essential structure for associative learning and behavioral memory in previous studies. 40 It will be interesting to investigate whether SARM1 deficiency improves learning and memory function after stroke.
In conclusion, our findings indicate that loss of SARM1 alleviates secondary thalamic neurodegeneration and improves somatosensory functional recovery after photothrombotic infarction. These favorable functional outcomes may be attributed to the attenuation of thalamic degeneration since the infarct volume was not reduced. Furthermore, loss of SARM1 was associated with the inhibition of autophagic overactivation. Autophagy may act downstream of SARM1 and contribute to secondary thalamic neurodegeneration. Anti-SARM1 therapy is a potential neuroprotective strategy for treating acute ischemic stroke.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from National Key R&D Program of China (2017YFC1307500), the National Natural Science Foundation of China (81200901), the Natural Science Foundation of Guangdong province of China (2017A030313575), the Medical Science Foundation of Guangdong Province of China (A2016268), the Southern China International Cooperation Base for Early Intervention and Functional Rehabilitation of Neurological Diseases (2015B050501003), Guangdong Provincial Engineering Center For Major Neurological Disease Treatment, Guangdong Provincial Translational Medicine Innovation Platform for Diagnosis and Treatment of Major Neurological Disease, Guangdong Provincial Clinical Research Center for Neurological Diseases.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
ZK and TY completed the experiments and wrote the manuscript. ZGF, LJJ and XSH participated in the animal experiments and Western blot. CXR, WJL did the image analysis and art work. LG and FYH imparted the animal modeling technics. ZJ and ZJS designed and funded the whole study.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.