
Abstract
The CAG/CAA expansion encoding polyQ huntingtin (mutant huntingtin [mHTT]) causes Huntington’s disease (HD), which is characterized by atrophy and loss of striatal medium spiny neurons (MSNs), which are preceded by neuropathological alterations in the cortex. Previous studies have shown that mHTT can spread in the brain, but the mechanisms involved in the stereotyped degeneration and dysfunction of the neurons from the striatum to the cortex remain unclear. In this study, we found that the mHTT expression initially restricted in the striatum later spread to the cortical regions in mouse brains. Such transmission was diminished in mice that lacked the striatal-enriched protein Ras-homolog enriched in the striatum (Rhes). Rhes restricted to MSNs was also found in the cortical layers of the brain, indicating a new transmission route for the Rhes protein to the brain. Mechanistically, Rhes promotes such transmission via a direct cell-to-cell contact mediated by tunneling nanotubes (TNTs), the membranous protrusions that enable the transfer of mHTT, Rhes, and other vesicular cargoes. These transmission patterns suggest that Rhes and mHTT are likely co-transported in the brain using TNT-like cell-to-cell contacts. On the basis of these new results, a perspective is presented in this review: Rhes may ignite the mHTT transmission from the striatum that may coincide with HD onset and disease progression through an anatomically connected striato-cortical retrograde route.
Keywords
INTRODUCTION
Wild-type huntingtin (wtHTT) is a ubiquitously expressed protein whose polyglutamine tract is encoded by the HTT CAG/CAA repeat expansion (mutant huntingtin [mHTT]) that causes Huntington’s disease (HD). HD results in the degeneration of D2 receptor-medium spiny neurons (D2R-MSNs), followed by D1R-MSNs in the striatum, and disrupts motor cognitive and psychiatric functions [1–3]. Prior to disease onset, striatal atrophy occurs, with a subsequent loss of cortico-striatal white matter (WM) connections in layer V [4]. As HD progresses, the disease affects other parts of the brain and the peripheral tissues [5, 6]. The mechanisms underlying the development of HD are not completely understood and likely several, but significant progress has been made over the past decades. For example, proteolytic processing, huntingtin aggregates, transcriptional dysregulation, energy metabolism deficits, synaptic dystrophy, oxidative stress, and inflammation [7–16] have been implicated as major contributors to HD development. However, their role in the precise orchestration in neuronal vulnerability is unclear. Further elucidation of the mechanisms governing neuronal vulnerability in HD should provide an opportunity to develop novel and effective therapeutic interventions. Herein, we review recent research works on Ras-homolog enriched in the striatum (Rhes) as a major driver of mHTT transmission and pathology in the brain.
CELL-TO-CELL mHTT TRANSMISSION
The first evidence that mHTT undergoes intercellular transmission emerged from the discovery of mHTT transmission in genetically normal and unrelated allografted fetal tissues in the striata of patients with HD [17]. In Drosophila, the movement of mHTT (exons 1–12 with Q138) was demonstrated from olfactory receptor neurons to other parts of the brain, coinciding with neuronal death [18, 19]. This movement requires intermediate glial cells to facilitate the nucleation of wtHTT exon 1 (Q25) [20]. Thus, mHTT shows an ability for prion-like propagation, a phenomenon that is now widely recognized [20–26]. Moreover, implantation of human HD cells harboring mHTT exon 1 (Q72–Q180) in the mouse cortex was later found in the striatum associated with MSN degeneration [27]. These results indicate that cell-to-cell mHTT transmission is widely observed across different species, including humans. Prior to HD onset, striatal atrophy and loss of cortico-striatal WM occurs in patients with HD, with a subsequent cortical cell death occurring in the later disease stages [4, 28–30]. Despite these studies, the mechanisms or role of mHTT transmission in mediating HD brain pathologies remains unclear. Moreover, whether the spread of mHTT from the vulnerable striatal region contributes to stereotypical loss of neurons in HD remains to be elucidated [31, 32].
ROLE OF RHES IN HD
Rhes (266 aa) belongs to the Ras GTPase family and is highly expressed in the MSNs of the striatum, and its expression is physiologically induced by thyroid hormone and dopamine innervations [33–35]. Rhes mRNA is also found to some extent in the cortex and hippocampus. In the striatum, the Rhes expression coincides with postnatal striatal maturation (P6–P25) in both D1R and D2R MSNs [36], indicating a potential role of Rhes in the developmental regulation of MSN function and maturation. We determined that Rhes is an atypical GTPase (1–171 aa) with small ubiquitin-like modifier (SUMO) E3-like (171–266 aa) functions [37, 38] and contains closely spaced SUMO post-translational modifications (PTM) on alpha helix surface-exposed lysine (110, K114, K120, and K124; Fig. 1) [39], similar to other SUMO E3-like proteins such as PIAS1 and RanBP2 [40, 41], and physiologically controls the SUMOylation and transcription of selected genes [39, 42]. We found that Rhes plays a critical role in the PTM SUMO modification, stabilization, and toxicity of the pathogenic form of mHTT in HD [42–44]. We demonstrated that Rhes interacts with mHTT in a farnesylation domain-dependent manner and promotes cellular toxicity by increasing the soluble forms of mHTT via SUMO1 modification [42], concurring with an independent study [45]. We also confirmed that Rhes or SUMO1 deletion diminishes mHTT toxicity and ameliorates HD-like behaviors in mouse models [44, 47]. The toxic role of Rhes in HD has been demonstrated in independent studies in various in vitro and in vivo models [45, 48–54]. These data indicate that Rhes-SUMO signaling plays a significant role in HD pathology, but the underlying mechanisms remain not fully understood. Our recent finding showed a great potential for Rhes as a vehicle for the intercellular spread of mHTT, a process that might facilitate the dissemination of HD pathology to other brain areas through tunneling nanotubes (TNT)-based communication routes.
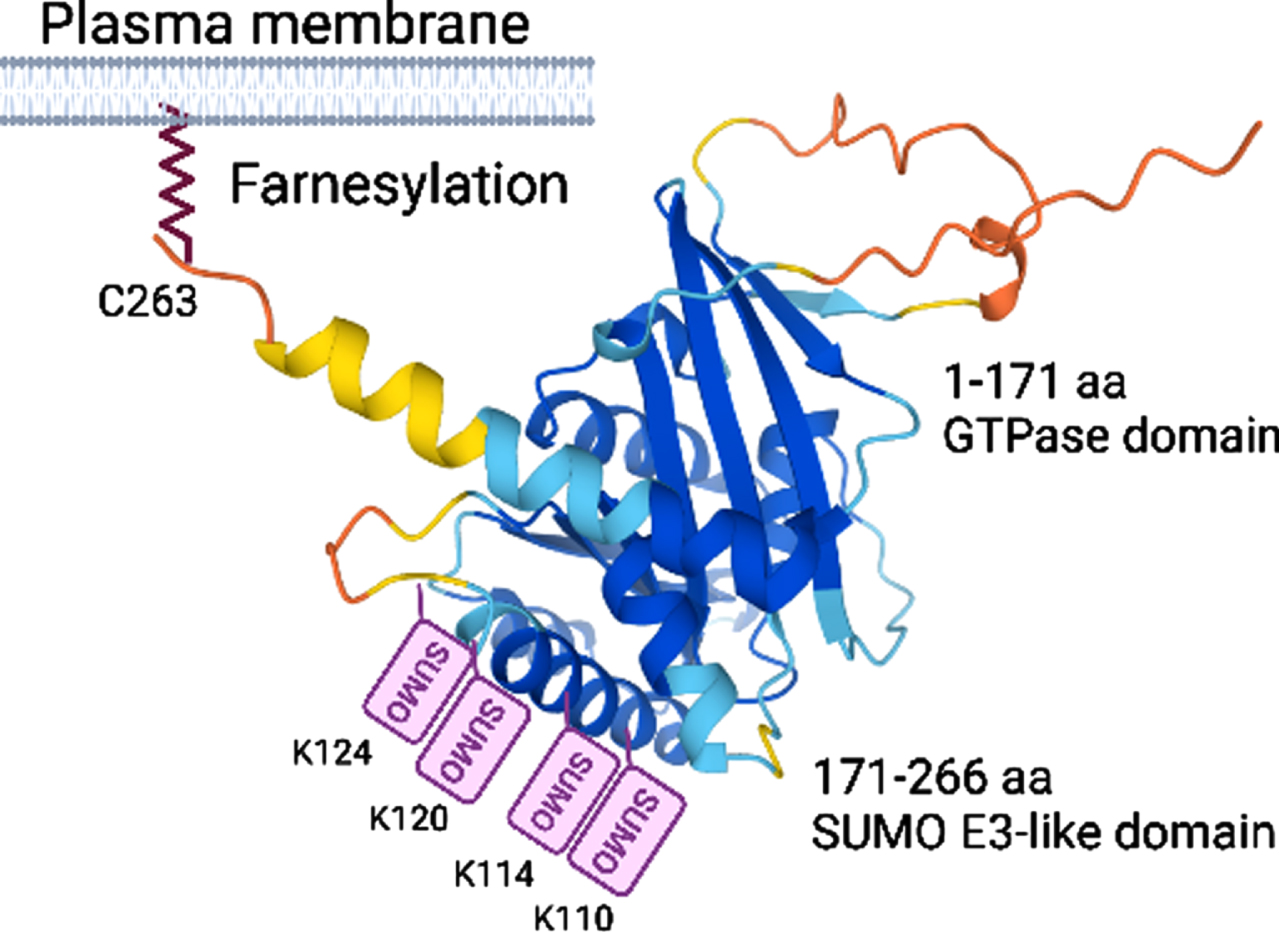
Alphafold structure prediction of Rhes, known domains, and PTM sites.
TUNNELING NANOTUBES
Cell-cell communication is fundamental for an organism’s development and for the maintenance of normal tissue structures and functions. TNT-like structures are predominantly actin-containing thin membrane protrusions emerging as essential mediators of direct cell-cell communication coordinating cellular processes such as electrical coupling, calcium signaling, small-molecule exchange, and organelle transfer [55, 56]. TNT-like thin structures are dynamic and fragile, do not adhere to the substrate in culture, and are destroyed by chemical fixation [57, 58]. Despite the advances in the demonstrations of the biological relevance of TNT-like cell-cell communication in development [59–61], immune response [62–64], neurovascular coupling [65, 66], and pathogenic protein spreading [55, 67–74] over the past decades, the mechanisms or its role in the spread of disease pathology remain unclear.
TNTs AND PATHOGENIC PROTEIN SPREAD
TNTs are demonstrated to be efficient and convenient means for the transmission of neurodegenerative disease proteins between cells. They serve as the major route for the transmission of infectious prions to non-infected cells [75]. TNTs also spread alpha synuclein and amyloid proteins between cells [76–79]. Similarly, the polyglutamine-expanded ATXN1 protein (ATXN1[82Q]), DNA-binding protein of 43 kDa (TDP-43), and Tau could transmit intercellularly via TNTs [80–84]. Although cellular models played a key role in discovering pathogenic protein transfer via TNTs, recent advancements in imaging technologies have identified TNT-like transfer of disease protein in vivo [73]. Thus, despite the likely critical roles of TNT-like mechanisms in pathogenic protein transmission, the molecular regulations remain largely unknown.
RHES PROMOTES TNT-LIKE COMMUNICATION AND MOVES BETWEEN MSNs IN THE BRAIN
While studying endocytosis, we serendipitously discovered that Rhes promotes TNT-like communication between neuronal cells and is self-transported from donor cells to acceptor cells [85]. Mechanistically, Rhes promoted TNT-like communication between cells through a membrane-binding mechanism [85]. Mutation of the prenylation domain (CAAX) cysteine 263, which releases Rhes from the cell membrane, failed to promote TNT-like communication [85]. Rhes can promote one or more thin and long TNT-like protrusions between cells (Fig. 2). While the GTPase domain is defective (1–171 aa) in producing TNT-like protrusions, the SUMO E3-ligase domain (171–266 aa) [37] induced TNTs but diminished Rhes transport to neighboring cells [85]. These results suggest that Rhes has distinct domains that differ in their ability to promote TNT- like protrusion formation and transport of protein cargo [85]. TNTs were first demonstrated in neuronal PC12 cells [86]. Depleting endogenous Rhes using CRISPR/Cas9 tools in PC12 cells [35, 87] reduced TNT formation, indicating that Rhes physiologically promotes TNT-like communications between cells [88].

Rhes promotes TNT-like cellular protrusions in striatal neuronal cells. (A) Striatal neuronal cells (STHdhQ7/Q7) expressing GFP-Rhes. The arrow shows the TNT-like protrusion, single (A) or multiple TNTs (B).
Beyond carrying Rhes protein as a cargo, Rhes-TNTs also transfer vesicular loads such as endosomes and lysosomes [85]. However, the identity of the Rhes-TNT cargo spectrum (proteins and vesicles) and the signals that mediate their entry into TNTs remain poorly understood. For example, live-cell imaging revealed that Rhes-TNT can also transfer caveolin 1, a protein component of caveolae (Fig. 3), indicating a role for Rhes in cell-to-cell vesicular microdomain signaling. By sharing these cargoes, Rhes-mediated TNTs can alter the functions in the neighboring and distant acceptor cells [71, 85]. Our preliminary evidence suggests that Rhes may affect mitophagy by interacting with the Nix mitophagy receptor in adjacent neuronal cells [89].
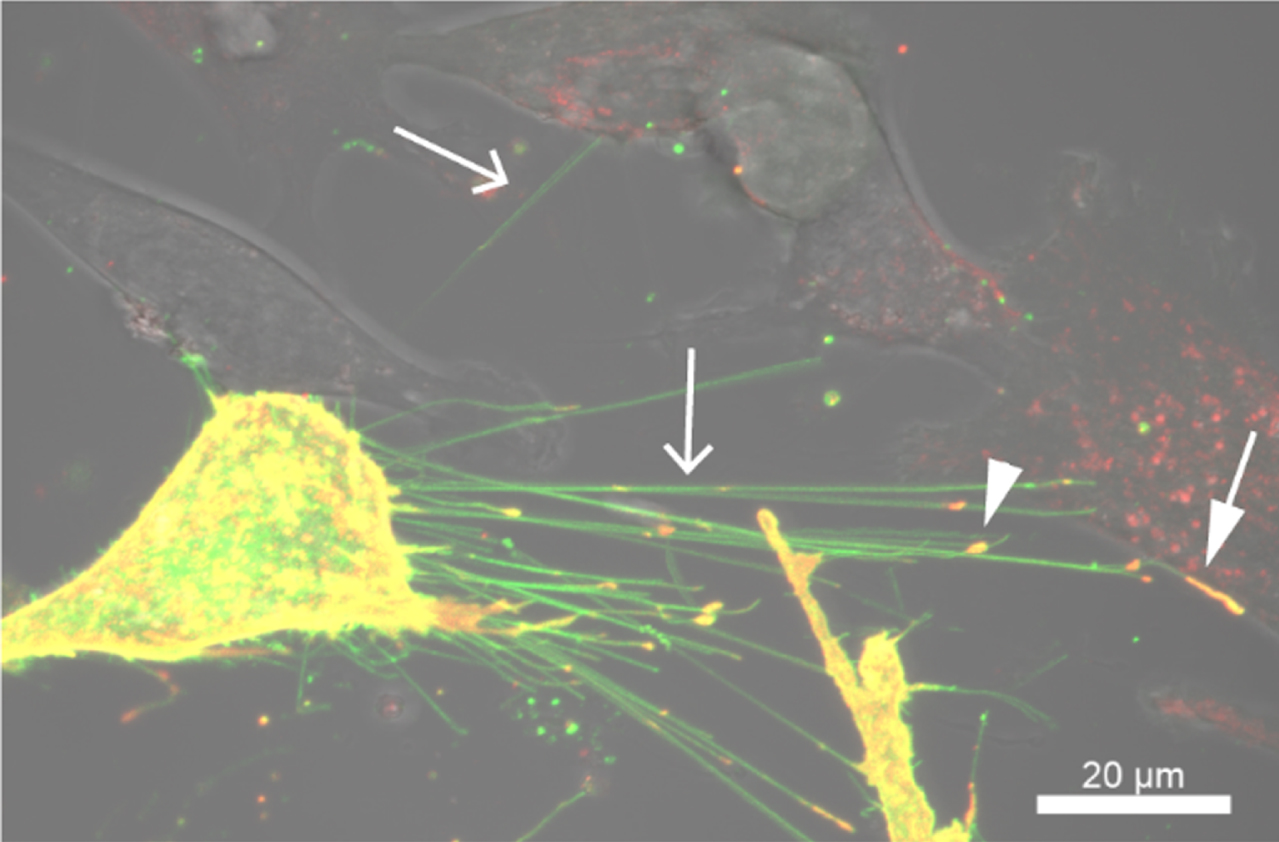
Rhes promotes TNT-like cellular protrusions in striatal neuronal cells. Confocal and DIC images of striatal cells expressing EGFP-Rhes and mCherry-tagged caveolin-1. Arrow indicates TNT, the arrowhead indicates caveolin-1 in the TNT, and the closed arrow indicates caveolin-positive TNT in the adjacent cell.
Thus, cell culture studies have shown that Rhes promotes TNTs and is transported to neighboring cells. However, determining whether a similar pattern of Rhes transport occurs in vivo is a prerequisite to establish its functional relevance. Therefore, by developing a strategy of cell-type-specific labeling using an AAV FLEX-switch delivery system for selective Cre-dependent (Cre-“On”) expression in MSNs, we made multiple critical observations. First, we found that Rhes is a novel inducer of TNT-like structures in MSNs [85, 88]. Next, we found that the Cre-On EGFP-Rhes expression produced multiple neuronal protrusions from D1R-MSNs that interacted (arrow) and Rhes puncta colocalized (arrowhead) with D1RtdTomato-MSNs, compared with Cre-On EGFP alone (Fig. 4). Third, we confirmed that Rhes transport between MSNs in an ex vivo three-dimensional model, an organotypic cortico-striatal brain slice culture that resembles the complexity of in vivo tissue [88]. Fourth, we found that Rhes expression restricted in the D1R-MSNs of the striatum was later found in the D2R-MSNs and in a subpopulation of cortical neurons in layer V and VI). Taken together, these results confirm that Rhes could move between MSNs within the striatum and from MSNs to the cortex in the mouse brain. Moreover, beyond neuromodulation via chemical and electrical synapses, these data provide understanding of how the protrusion-based communication subserves non-classical signal transmission between different cell types in the striatum. Our data show that Rhes is critical for mHTT spread through cell-to-cell TNT-like contact in vitro and in vivo.
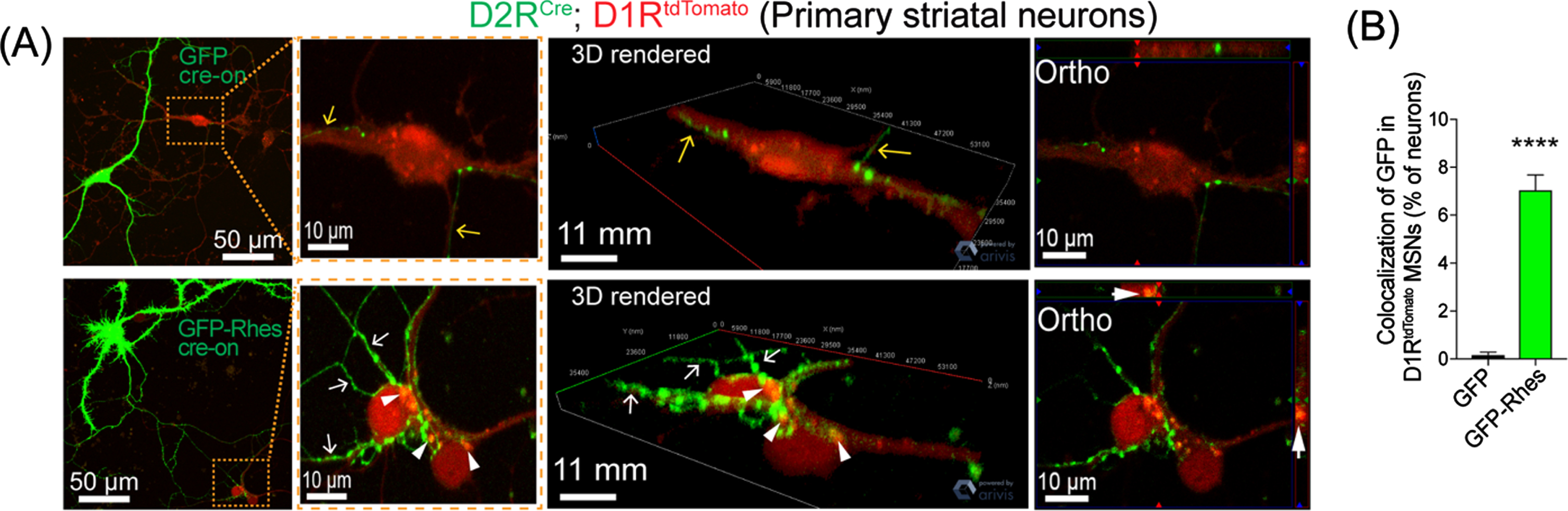
Rhes travels from D2R MSN to D1R MSN in vitro. (A) confocal live-cell images and insets of MSN culture from D2RCre; D1RtdTomato mice infected (DIV5) with AAV-PHP.eB Cre-on GFP or AAV-PHP.eB Cre-on GFP-Rhes (MOI 5). Yellow and white arrow show neuronal processes of GFP or GFP-Rhes from Cre(+) D2R MSN, respectively. The arrowhead shows GFP-Rhes puncta colocalization in Cre(–) D1RtdTomato MSN. 3D rendering and orthogonal (ortho, single plane) images show GFP-Rhes puncta inside the D1RtdTomato MSN. (B) Bar graph shows quantification of colocalization of GFP signal in D1RtdTomato MSNs (% of neurons) for GFP and GFP-Rhes groups (n = 16 D1RtdTomato MSNs/group). Data are mean±SEM; Student’s t-test, ***p < 0.0001.
RHES MEDIATES CELL-TO-CELL mHTT TRANSPORT VIA TNTs
As described earlier, Rhes is associated with mHTT and HD pathology [42, 48–53], so we considered that Rhes might carry mHTT via the TNT-like protrusions in striatal neuronal cells. Low signal intensity of endogenous FL-wtHTT (detected with MAB2166 antibody) was found in Rhes-induced TNTs [85], which were readily positive for endogenous FL-mHTT. Similarly, Rhes-induced TNTs also contained the N-terminal toxic fragment HTT exon1-2 89Q (HTT N171-89Q) (Fig. 5) but little HTT N171-18Q [85]. FACS/coculture data showed that HTT N171-89Q alone is not transported between cells; however, Rhes enhanced mHTT transport manyfold [85]. In primary striatal neurons, GFP-Rhes TNT-like protrusions were colocalized with mHTT in neighboring cells [85], indicating transmission of mHTT by Rhes between neurons.
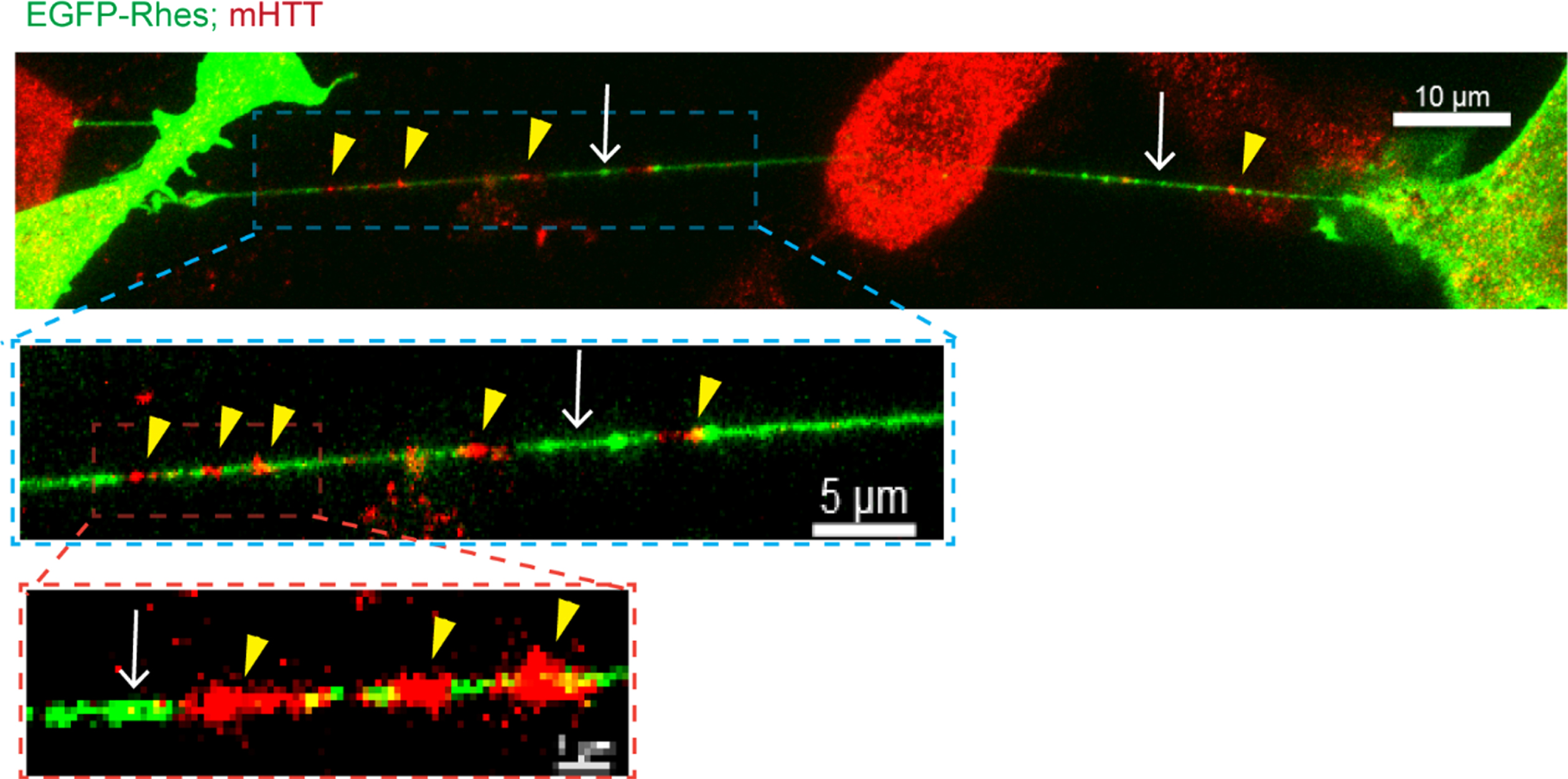
Rhes transports mHTT via TNT-like protrusions. Confocal images of EGFP-Rhes in mutant striatal cells (STHdhQ111/Q111) with immunocyto-chemistry for endogenous FL mutant HTT with anti-HTT MAB2166 antibody (Red). The arrow shows the TNT-like protrusion. Arrowhead shows mHTT in the TNTs.
Rhes requires farnesylation for toxicity and mHTT SUMOylation [42, 45]. We used a farnesylation-defective Rhes C263S mutant and determined its role in Rhes-mediated mHTT transport. Fluorescence-activated cell sorting analysis/coculture experiments revealed that the Rhes C263S mutant almost completely failed to transport mHTT to the acceptor cells, similarly to the effect of the actin-polymerization inhibitor, cytochalasin D. Moreover, mHTT transport in SUMO-depleted cells and SUMOylation-defective mHTT (HTT N171-79Q K/R) transport by Rhes were significantly diminished [42, 85]. Taken together, the results of these studies indicate that Rhes promotes TNT-like protrusions and actin- and SUMO-based movements of cellular cargoes and itself to distant cells. Although the exact mechanisms used by Rhes-TNTs for mHTT delivery into the acceptor cell are unknown, our findings from the live-cell imaging analysis implicated endocytosis-like mechanisms [71, 85].
RHES PROMOTES mHTT TRANSPORT IN THE BRAIN
We also examined whether mHTT can be transported by Rhes in the intact brain. We prepared lentiviral (LV) bicistronic vectors expressing EGFP and mCherry HTT containing HTT N171-18Q (LV-wHTT) or HTT N171-89Q (LV-mHTT), separated by the P2A sequence. In this scenario, EGFP and HTT are made by one mRNA as two separate proteins, thereby allowing the monitoring of EGFP/mCherry fluorescence. We stereotaxically injected LV-wtHTT or LV-mHTT unilaterally in the striatum of 3-month-old adult WT and Rhes–/– (Rhes knockout [KO]) mice. After 2 months, the brains were fixed and processed for EGFP and mCherry fluorescence detection by confocal microscopy. As expected, we observed EGFP and mCherry (HTT) expressions at the injection site in the striatum (Fig. 6A). By contrast, mCherry-mHTT intensity was detected 500 μm away from the injection site in the WT mice, indicating mHTT movement within the striatum (Fig. 6B). The signal was markedly reduced in the Rhes KO mice (Fig. 6B). The wtHTT underwent little transport in the striatum compared with the mHTT, and the WT and Rhes KO mice showed no significant differences [88]. A strong mHTT intensity was observed in the cortex (Layer V) of the WT mice and was again diminished in the Rhes KO mice (Fig. 6C) [88]. Most mHTT was distributed perinuclearly in the striatal and cortical cells of the brains of the WT and Rhes KO mice. These results imply that the mHTT protein is transported within and from striatal cells to cortical areas and that Rhes is a major driver of this transport [88]. Prior evidence hints that mHTT is expressed in exosomes [90]. However, we confirmed that Rhes is not detectable in exosomes [88]. Thus, Rhes is a major driver of mHTT transmission, likely via a TNT-like mechanism [85, 88].
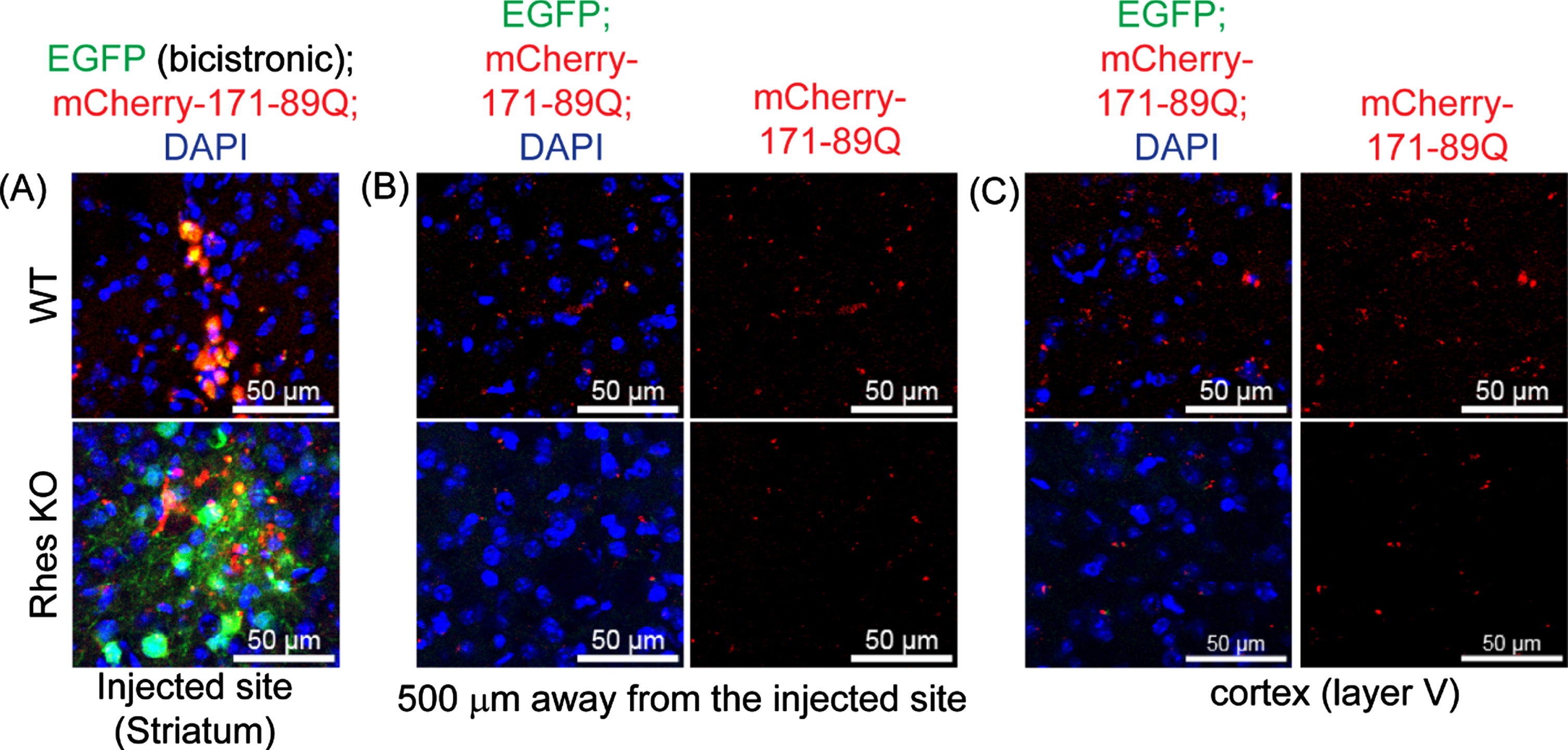
Rhes promotes mHTT transmission in the brain. Representative confocal image of brain slices from WT and Rhes KO (4 moths old) expressing bicistronic expression of EGFP (green) and the mHTT 171-89Q (mcherry, red) and cell nuclei stained with DAPI (blue) in the injected site (A), 500μm away from the injected area (B), and in the cortex (layer V) (C).
CONCLUSIONS AND FUTURE PERSPECTIVES
Finding that Rhes mediated mHTT transmission between cells can potentially have the most significant impact in understanding the spread of HD pathology. Although the mechanisms are still unknown, an integrated conceptual framework would be useful for understanding how such spreading from the striatum is initiated and extends to other brain regions. From a therapeutic perspective, it is critical to stop the development of the processes that cause mHTT to spread. These mechanisms are anticipated to occur early in the course of the disease. Therefore, for effective therapeutic strategies, a detailed understanding of the intricate biology of mHTT spreading is essential.
We propose a three-step conceptual framework in which the Rhes in the striatum may ignite the process of mHTT spreading. We argue that the three steps differ with respect to the cellular status and function of the mHTT (Fig. 7): posttranslational SUMO modification, aggregates and inclusions in MSNs, and key abnormalities of the protein quality control system, including autophagy and ubiquitin proteasome pathways. While SUMO modification of mHTT by Rhes and other E3 ligase such PIAS1 shifts the mHTT equilibrium from an insoluble to a soluble state, downstream cellular dysfunctions play a critical role for the initiation of mHTT protein spread. We hypothesized that soluble mHTT will interfere with the protein quality control system forming protein aggregates and diverse oligomeric species. This enhanced the abundance and accumulation of the mHTT species saturate the protein quality system and will result in homeostatic disturbance in MSNs and advancement of HD pathology. Our work suggests that the gradual spread of HD pathology from cell to cell and region to region in a stereotypical manner is facilitated by Rhes-mediated spreading of pathological mHTT via TNT-like routes. Targeting Rhes via small molecules or genetic strategies may offer significant protection in HD. Thus, our study contributes to the improved understanding of how HD may spread through anatomically connected cortico-striatal pathways in the brain.
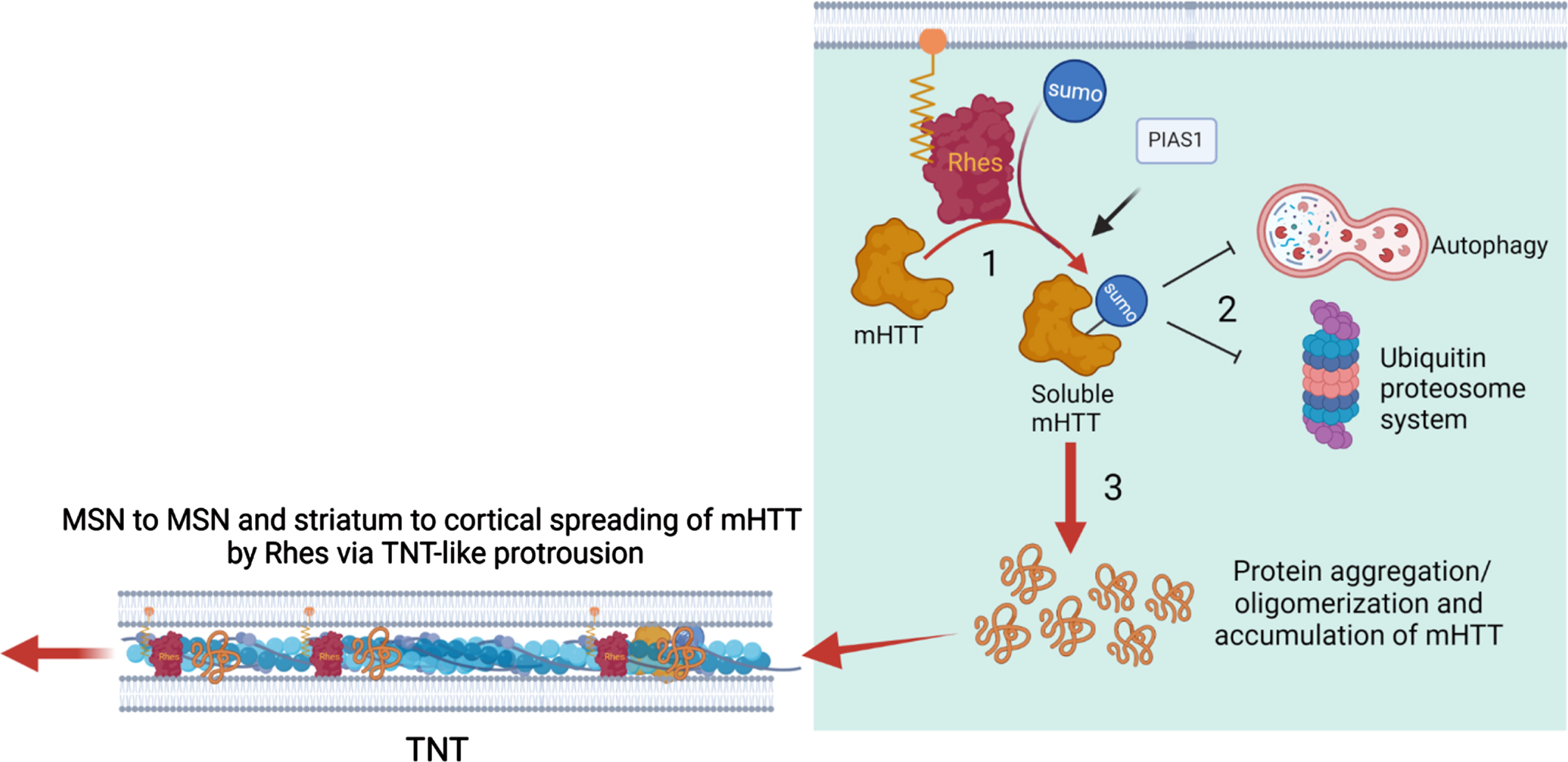
Working model for Rhes-mediated cell-cell mHTT transmission from the striatum. See texts for details.
Footnotes
ACKNOWLEDGMENTS
The author would like to thank Dr. Manish Sharma and Dr. Uri Nimrod Ramírez-Jarquín for the image acquisition at the Max-Planck Institute of Neuroscience, Jupiter, Florida. Research that led to the publication of the review article was supported by grant awards from NIH/NINDS R01-NS087019-01A1, NIH/NINDS R01-NS094577-01A1, and partly from the Cure for Huntington Disease Research Foundation (CHDI).
CONFLICT OF INTEREST
The author declares that there are no competing interests associated with this manuscript.