
Abstract
Huntington’s disease (HD) is a fatal, inherited neurodegenerative disorder caused by a mutation in the huntingtin gene (HTT). While mutant HTT is present ubiquitously throughout life, HD onset typically occurs in mid-life, suggesting that aging may play an active role in pathogenesis. Cellular aging is defined as the slow decline in stress resistance and accumulation of damage over time. While different cells and tissues can age at different rates, 9 hallmarks of aging have emerged to better define the cellular aging process. Strikingly, many of the hallmarks of aging are also hallmarks of HD pathology. Models of HD and HD patients possess markers of accelerated aging, and processes that decline during aging also decline at a more rapid rate in HD, further implicating the role of aging in HD pathogenesis. Furthermore, accelerating aging in HD mouse and patient-derived neurons unmasks HD-specific phenotypes, suggesting an active role for the aging process in the onset and progression of HD. Here, we review the overlap between the hallmarks of aging and HD and discuss how aging may contribute to pathogenesis in HD.
Keywords
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant, progressive and eventually fatal neurodegenerative disease caused by an expanded polyglutamine-encoding CAG tract in the Huntingtin (HTT) gene, which gets translated into a mutant HTT (mtHTT) protein [1]. mtHTT disrupts several processes in the cell, eventually leading to dysfunction and death of the caudate and putamen, and the cerebral cortex. HD is characterized by psychiatric, cognitive, and progressive movement abnormalities [2]. Age-of-onset of disease is negatively correlated with the number of CAG repeats [3–6], with motor symptoms beginning between ages 35–45 and death occurring 10–20 years after motor-onset [2]. However, the number of repeats only partially explains age-of-onset; individuals with identical tract lengths can have disease onset many decades apart [7]. This suggests that both genetic and environmental factors outside the CAG repeat can influence disease pathogenesis.
Although there is evidence which suggests that neurodevelopmental abnormalities can occur in adult-onset HD [8, 9], those with the mutation that causes adult-onset HD can live several decades without overt symptoms. This suggests that aging, particularly in adult-onset HD, may play a role in pathogenesis. However, how aging may contribute to HD pathogenesis is controversial. Two primary hypotheses have arisen: (1) Aging passively contributes to HD by the simple passage of time, where mtHTT-induced microdamage accumulates over many decades before reaching a threshold of disease onset, or (2) events that occur during the aging process sensitize neurons/the brain to the mtHTT insult and actively contribute to disease onset and/or progression.
Aging is a risk factor for many diseases, including neurodegenerative disease [10]. While chronological aging is defined as the number of years an organism is alive, biological aging describes the overall health of the organism [11, 12]. Biological aging is defined by 9 hallmarks: telomere attrition, epigenetic alterations, altered intercellular communication, dysregulated nutrient sensing, loss of proteostasis, mitochondrial dysfunction, genomic instability, stem cell exhaustion, and cellular senescence [13]. While both chronological and biological age are risk factors for disease, biological aging seems to play a larger role in disease burden of many age-related diseases [14–16]. This suggests that processes that decline during aging play an active role in disease. In HD, many of these aging hallmarks are also hallmarks of disease pathogenesis. Here, we discuss the evidence for accelerated aging in HD, the overlap between the cellular hallmarks of aging and the cellular pathogenesis of HD, and how aging may exacerbate toxicity in HD.
EVIDENCE FOR ACCELERATED AGING IN HUNTINGTON’S DISEASE
Telomere attrition
Telomeres are repetitive TTAGGG nucleotide sequences that act as a cap at the end of chromosomes that insulate them from attrition during replication [17]. Telomeres are maintained by telomerase, an enzyme that is relatively silent throughout the body shortly after birth. Thus, telomeres undergo successive shortening with age, and biological aging has been linked to shortened telomeres [18]. In addition to biological age, shortened telomeres are associated with several diseases, including mood disorders, cancer, type 2 diabetes, and chronic obstructive pulmonary disease [19–22]. While telomere attrition may not occur is post-mitotic neurons [23], several neurodegenerative diseases have shortened telomeres in leukocytes compared to age and sex-matched control individuals [24, 25]. Interestingly, of all neurodegenerative diseases, HD leukocytes have the shortest telomeres [24]. This was also shown in a follow-up study [26], and could potentially be used as a peripheral blood biomarker for HD progression [27, 28]. In addition, there is an association between age-of-onset of HD and SNPs that influence telomere length [29], further demonstrating an association between telomere shortening, a marker of advanced biological aging, and HD.
Epigenetic alterations
The epigenetic landscape of DNA and histones changes with age. Widespread DNA hypomethylation and loss of heterochromatin have been described [30]. In addition, in the aging brain, distinct patterns of epigenetic and, consequently, transcriptional changes occur. In this way, the epigenetic landscape of DNA and chromatin has come to be a predictor of biological age [31–33]. In the brains of HD patients, accelerated epigenetic aging has been shown in the striatum, the most affected part of the brain in HD [34]. Taken together with data from telomeres in HD, this demonstrates that HD brains also have accelerated biological age compared to age-matched controls. This is in concordance with the findings of others that show accelerated brain epigenetic aging in other neurological disorders, including Alzheimer’s disease, Down’s syndrome, and HIV-associated neurocognitive disorders [35–37].
OTHER HALLMARKS OF AGING INVOLVED IN HUNTINGTON’S DISEASE PATHOGENESIS
Altered inter- and intra-cellular communication and stress signaling
Intercellular communication refers to the non-autonomous signaling between cells and tissues, whereby one tissue can have an effect on the other. One of the most common examples of intercellular communication is inflammation. Immune cells such as neutrophils, B cells, T cells, macrophages, astrocytes and microglia release inflammatory signals to surrounding cells as a defense system against cellular harm [38]. Additionally, non-immune cells under stress can release cytokines and chemokines to surrounding cells to boost defenses against foreign pathogens [39]. Together, this response is meant to be acute and resolved by subsequent release of anti-inflammatory factors and tissue remodeling [40]. Throughout aging, however, pro-inflammatory signaling is elevated, and anti-inflammatory factors become diminished, leading to chronic inflammation, deemed “inflamm-aging.” For a detailed review of inflamm-aging pathways, we refer the reader to [41].
Reactive microglia have been found in HD brains that correlate with HD pathology [42, 43]. Macrophages, monocytes, and microglia isolated from YAC128 and R6/2 mice as well as from peripheral blood of HD patients are hyperactive upon stimulation [44, 45], similar to the stimulus-induced hyperactivity observed in microglia from aged mice [46]. Astrocytes can also be dysfunctional in HD brains, with a decreased ability to take up glutamate and increased glutamate release [47, 48]. Interestingly, glial cells from the white matter of adult brains possess shorter telomeres than grey matter glial cells [23]. This could contribute to the observed white matter loss in HD patients prior to grey matter loss, and may suggest that “older” glial cells contribute to HD pathology at earlier stages. Pro-inflammatory markers such as IL-6 and matrix metalloproteinase 3 (MMP3) are also elevated in the plasma and CSF of HD patients, respectively [44, 45], suggesting increased systemic inflammation in HD. Additionally, in mice with mtHTT expressed only in microglia, there is an increase in neuronal cell death [49]. Moreover, reducing mtHTT in astrocytes slows progression of phenotypes in BACHD mice [50], further providing evidence for the role of non-autonomous contributions from glia to HD pathology.
In addition to glial and peripheral immune cell contributions to inflammation, several intracellular pathways can also lead to increased inflammation and cellular dysregulation in both aging and HD. Elevated levels of reactive oxygen species (ROS), which we discuss in greater detail below, can chronically activate nuclear factor kappa B (NF-κB), a transcription factor that induces expression of pro-inflammatory genes [51]. RNA sequencing of HD patient brain found upregulation of several NF-κB target genes [52], and inhibition of NF-κB signaling in astrocytes is sufficient to delay phenotypes in R6/2 mice [53]. Mammalian sterile 20-like kinase1 (MST1) was also found to be activated in post-mortem HD cortical tissue and CAG knock-in Q111 mice [54]. MST1 promotes the innate immune response, increasing inflammation [55]. Furthermore, wildtype HTT is a stress response protein, serving as a scaffold protein for the recruitment of DNA damage response proteins and early endosomes [56–59]. mtHTT does not recover from these responses as quickly, resulting in aberrant signaling and associated stress responses, which can indirectly activate immune response pathways. Together, these aberrant stress signaling pathways coordinate not only to increase damage in HD, but can also cause damaging chronic inflammation.
Dysregulated nutrient sensing
In cells, the master regulator of nutrient sensing is insulin-like growth factor (IGF-1). IGF-1 is activated by glucose, in a similar manner to insulin, and this pathway, which is the most-conserved pathway in organisms, is referred to as the insulin and IGF-1 signaling (IIS) pathway [60]. In aging, IGF-1 declines and contributes to aging [61], although paradoxically, reducing activity of downstream effectors of IGF-1 such as mTOR, FOXO, or Akt promote longevity [62, 63]. This is hypothesized to be because of the slowed metabolism and growth of organisms with decreased IIS activity [64].
Dysregulated nutrient sensing also occurs in HD patients. However, contrary to the hypometabolism often seen in the elderly, patients with HD most often exhibit hypermetabolism, with high rates of weight loss despite an increase in caloric intake [65]. FOXO, mTOR, and Akt, all downstream effectors of the IIS pathway are dysregulated in HD patient brains and models of HD. Activation of the IIS pathway via modulation of FOXO, mTOR, or Akt is protective in HD models by ameliorating mitochondrial function and metabolic rates [66–71]. Somewhat paradoxically, although IIS activation is protective in HD models, intermittent fasting, which reduces IGF-1 levels, promotes clearance of mtHTT in YAC128 mice [72]. To support this, compounds such as rapamycin, which blocks mTOR activity, and metformin, which inhibits IIS, promote autophagic clearance of mtHTT and improve phenotypes in HD mice [73–75]. Furthermore, plasma IGF-1 levels were found to be positive predictor of cognitive decline in HD patients [76]; thus, how exactly the IIS pathway can be protective in HD is still debated.
Loss of proteostasis
Proteostasis, or ‘protein homeostasis’, refers to the process of quality control regulating protein abundance and conformation. The network of proteins involved in maintaining proteostasis include proteins involved in translation, molecular chaperones, which aid in re-folding misfolded proteins, as well as the ubiquitin-proteasome system (UPS) and lysosome/autophagy machinery, which degrade excess and damaged proteins [77, 78]. Canonical protein degradation involves tagging proteins with mono or poly-ubiquitin chains to be recognized by the proteasome or autophagosomes [79–81]. During the process of aging, the proteostasis network in the brain declines in function, evidenced by the appearance of protein aggregates even in aging brains without apparent neurodegenerative disease [82–84]. This is thought to be due, at least in part, to an increase in errors in translation and protein misfolding as a result of accumulated damage and subsequent oxidative and carbonylation of proteins [84].
Autophagosomes and lysosomes decrease in number and activity with aging in several tissues [85, 86]. Furthermore, inhibition of autophagy leads to premature aging phenotypes [87], and stimulating autophagy by caloric restriction and pharmacologic stimulation of upstream autophagy pathways promotes longevity in nearly all model organisms from C. elegans to rhesus monkeys [88]. Additionally, the UPS and lysosome/autophagy pathway become dysregulated with age, with shifts in the ubiquitylation of the proteome and a decrease in the activity of the proteasome and autophagic machinery, further contributing to the decline in protein integrity observed in the aging brain [77].
A decline in overall proteostasis has been linked to disease progression in models of HD [89, 90]. Aggregates of mutant HTT have been shown to sequester ubiquitin as well as components of the proteasome [91], which could result in decreased proteasome function, although this hasn’t directly been linked to cellular decline. It is possible that proteasome sequestration is dynamic, and may not occur at a high level all the time but may be enough to slow down the proteostasis network as a whole [92]. Interestingly, while primary cortical neurons from HD mice do not exhibit aggregation of mtHTT, we recently found that inducing aging in these neurons caused oligomers of mtHTT to form [93]. Additionally, the E3 ubiquitin ligase WWP1 was found to aberrantly ubiquitinate mutant HTT and impair degradation in an age-dependent manner [94], further suggesting a relationship between age and mtHTT toxicity.
Contrary to what is typically observed during aging, autopsied brains of HD patients have increased size and number of autophagic vesicles [95]. Additionally, staining of LC3II, a marker of autophagic vesicles, is increased in HD striatal neurons [96], and UBR5, a ubiquitin ligase responsible for proteosomal degradation is increased in HD iPSCs [97]. Despite this increase, overexpression of UBR5 is protective in HD iPSCs, suggesting either dysfunction in this pathway or an overwhelming of the proteosomal system in HD cells [97]. Indeed, despite this increase in vesicle size and number, actual flux of some proteins and organelles is decreased, possibly due to defects in cargo recognition [98]. wtHTT can function as a scaffold for autophagy [99], and partial loss of this function by mtHTT may contribute to autophagy defects. While the mechanism of autophagy impairment in aging and HD is slightly different, the result is similar: an accumulation of damaged proteins and organelles. One such organelle is the mitochondria, which are turned over by the autophagy/lysosomal pathway. Damaged mitochondria accrue in aged tissues as well as HD neurons, creating a feed-forward loop for both ROS damage and lysosomal impairment, which are particularly susceptible to ROS damage [100].
Mitochondrial dysfunction
There is extensive evidence for mitochondrial dysfunction in HD patients and in many models of HD. This was originally discovered by in vivo positron emission tomography (PET) imaging of brains of HD patients, where cerebral metabolism defects and atrophy were found [101]. These metabolic defects occurred early and severity was associated with functional decline in HD patients [102–105]. Follow-up studies in animal models confirmed altered metabolic rates and found distinct changes in mitochondrial complexes [106–111], although this is not always the case [112, 113]. Decreased cerebral glucose metabolism is also a characteristic of the aging brain [114–116].
Nicotinamide adenine dinucleotide (NAD+) is a metabolite essential for cellular function that has been shown to decline with age [117–119]. NAD+ can be synthesized de novo from its precursor tryptophan, through the Preiss-Handler pathway, or through the recycling of components from enzymes that consume NAD+ to carry out metabolic functions [120]. This process is dependent on nicotinamide phosphoribosyltransferase (NAMPT), an enzyme that converts salvaged nicotinamide to NAD+ [121]. The main causes of NAD+ decline in aging are a decrease in Nampt expression [122] and an increase in NAD+-dependent activity by the poly-(ADP-ribose) polymerases (PARPs), Sirtuins (SIRTs), and CD38 [123]. In line with this, exogenous nicatinamide upregulates BDNF, and PGC-1α gene expression and improves motor phenotypes in R6/1 HD mice [124]. PARPs are enzymes that create ADP polymer chains using NAD+, which occurs during DNA repair [125]. PARP activity, particularly PARP1 activity, is increased in aging tissue, possibly due to the accumulation of DNA damage [126]. This increased utilization of NAD+ by PARP1 is hypothesized to be the cause of the eventual reduction in SIRT activity [127]. PARP1 hyper-activity has also been observed in post-mortem HD brains [128]. In addition, inhibition of PARP1 activity is neuroprotective in R6/2 HD mice [129]. Similar to findings in aged brains, SIRT1 and SIRT3 expression are decreased in cultured HD neurons and HD brains [130–132], and SIRT1 and SIRT3 activation is beneficial HD neurons as well as R6/1 and YAC128 HD mice [130, 133]. While accelerated aging has not been directly linked to the observed changes in enzymatic activity of NAD+-consuming enzymes, the fact that levels change in a similar manner in both aging and HD is suggestive of an accelerated aging component to HD pathogenesis.
In addition to cellular metabolic defects, many models of HD as well as post-mortem brains from HD patients have shown defects in mitochondrial structure and function in similar ways to mitochondrion from the aging brain. Mitochondria are dynamic, networked organelles, undergoing fission or fusion with the network in response to changing cellular environments [134]. In the aging brain, there is decreased abundance in mitochondria and a change in shape toward smaller, rounded, and less-networked mitochondria in many cell types, including neurons [135–138]. In addition to changing structure, mitochondria from aged tissues have decreased oxidative phosphorylation and ATP production [139–141]. In HD, similar observations have been made in neuronal mitochondria. Abnormal mitochondrial dynamics and increased activity of the GTPase DRP1, responsible for mitochondrial fission, have been reported in HD models and post-mortem brains of HD patients [142–145].
Decreased calcium handling is characteristic of mitochondria from aging neurons, and is also observed in HD brains. Mitochondria take up calcium through the calcium uniporter, which helps to buffer calcium input in neurons [146]. Mitochondria from aging neurons do not do this effectively [147]. Calcium handling defects are also observed in transgenic HD mice and rats as well as lymphoblasts from HD patients [148–150], although this is not seen in some HD mouse neurons until they are challenged with NMDA [151, 152].
ROS are byproducts of cellular metabolism that are typically cleared by endogenous antioxidants in the cell [153, 154]. While ROS and antioxidants are typically in homeostatic balance in the cell, ROS can overwhelm antioxidant systems, causing damage to DNA, RNA, proteins, and organelles [155].Throughout the process of aging, this homeostatic imbalance can become pronounced. Antioxidant protein levels and activity decline [156, 157], and age-related disruptions in mitochondrial activity cause more ROS generation, and subsequent damage to DNA and biomolecules [158]. In a similar manner, HD neurons have decreased antioxidant activity, and increased ROS and ROS-induced damage [159–161]. However, recently, we found that in primary cortical neurons from humanized HD mice, the ROS-induced damage and hypersensitivity to oxidative stress observed in HD neurons is dependent upon biological age [93], suggesting that aging uncovers stress-induced phenotypes in HD. Moreover, through aging ROS can promote cellular senescence [162], and recently, RNA sequencing of brains from HD knock-in mice revealed that striata from symptomatic HD mice possess gene signatures associate with cellular senescence [163].
Cellular senescence
Cellular senescence refers to the cell cycle arrest of replicative cells due to aging-related factors [164]. Senescent cells exist in a pro-inflammatory state and can release cytokines, proteases, and growth factors collectively known as a senescence-associated secretory phenotype (SASP) [165]. While senescence in this sense doesn’t apply to terminally differentiated cells such as neurons, aged neurons have increased expression of pro-inflammatory markers, markers of senescence such as p53 and p16INK4A, tumor suppressors that can also trigger senescence-related transcriptional changes in aging, and an increase in senescence-associated β-galactosidase activity [166]. In HD, mtHTT can bind to p53 and increase protein activity [167]. p16 and p53 gene expression are also increased in the striata of HD knock-in mice [163]. Furthermore, both astrocytes and microglia from aged brains acquire SASPs [168, 169]. Furthermore, there is an increase in pro-inflammatory cytokines and matrix metalloproteinases such as MMP-9 in HD mice as well as post-mortem brains of patients with HD [170].
DNA damage and genomic instability
Genomic instability refers to the frequency of mutations within the genome of cells. Chromosomal changes and DNA mutation rate increase with age [171–174]. Mutations in aging are thought to be caused, at least in part, by failure to repair or improper repair of DNA damage. DNA damage continuously occurs in cells due to free radical damage and environmental agents, such as UV rays. DNA damage comes in several forms, including insertions/deletions/substitutions, bulky adducts, single-strand breakage and double-strand breakage [175–177]. Damage accrued independently of replication can be repaired by one of four known pathways, each with multiple scaffolds and polymerases [178]. While each DNA damage response (DDR) pathway has a canonical lesion to repair, there is significant crosstalk between pathways. For a comprehensive review on DDR pathways, see [178].
Throughout the course of aging, several forms of DNA damage can occur, with the oxidative lesion, 8-oxodeoxyguanine (8-oxo-dG), being one of the most prevalent [179–182]. 8-oxo-dG is repaired primarily through the base-excision repair (BER) pathway [182], although both nuclear excision repair (NER) and mismatch repair (MMR) can also repair oxidative lesions [183]. Repair of oxidative lesions is initiated by 8-oxoguanine glycosylase (OGG1), which removes the lesion [184]. The resulting gap in DNA triggers PARP1, which uses NAD+ to add poly(ADP-ribose) chains around the gap, recruiting polymerases to fill the gap [185]. This is important because, throughout the course of biological aging, there is evidence that OGG1-dependent activity can trigger the aging process [183]. Furthermore, aging-related increases in DNA damage and/or aberrant repair is hypothesized to be a cause of age-related increase in cancer risk. However, if too much damage occurs, cellular senescence or apoptosis can be triggered [186].
While there is currently no evidence for a change in overall genomic instability in HD [187, 188], nuclear DNA modifications and increased double-stranded breaks have been observed [189, 190], and there is somatic instability of the CAG repeat itself. Additionally, there is evidence for disrupted nuclear pore complexes in HD [191], which also occurs in aging [192]. Breakdown of the nuclear pore complex can cause excessive DNA damage and aberrant intracellular communication, another hallmark of aging [193, 194]. These events combined may contribute to or cause instability in HD neurons.
There is an inverse relationship between somatic instability of the CAG repeat and age-of-onset of disease [195–197]. Instability may even be necessary in the brain for pathogenesis [197–199]. This instability seems to be dependent upon DNA repair pathways; crossing mouse models of HD with mice deficient in MMR (Msh2, Msh3, Mlh1, or Mlh3 –/–mice) or BER (Ogg1 or Neil1 –/–mice) attenuates instability of the CAG tract and delays HD-relevant phenotypes [198, 201]. Furthermore, a GWAS performed on tissue from HD patients found that several SNPs which influence age-of-onset of HD were found at or near genes involved in the DDR [202]. In a follow-up GWAS investigating genetic modifiers of HD progression, there was a significant signal in MSH3, an MMR protein [203]. This provides further evidence for the role of DNA damage-associated pathways in HD. Recent work from our group demonstrated that accelerating biological age of HD mouse neurons or brains or neurons derived from HD patient iPSCs results in a dramatic and selective increase in DNA damage that can be exacerbated by cellular stress [93], although it is still currently unclear whether age-associated DNA damage can influence instability. Some DDR pathway proteins show a decline in efficiency with age [204], leaving the possibility that aging contributes to somatic expansion by creating heightened, but inefficient DDR pathways. Alternatively, somatic expansion could be triggered as a direct result of increased DNA damage and associated increases in repair, before age-associated declines in DDR efficiency. Both increased DNA damage and inefficient DDR pathways, specifically in the MMR pathway, contribute to microsatellite instability in aging [205]. However, how these pathways change in the brain with aging is still unknown; therefore, how exactly this hallmark may contribute to somatic instability in HD is still unknown and of great importance to HD research.
CONCLUSION AND FUTURE PERSPECTIVES
Many of the hallmarks of aging are also mechanisms of pathogenesis in HD. While all these processes decline with age, they seem to decline more rapidly in patients with HD, suggesting a synergistic interplay. In many adult-onset neurodegenerative diseases, the role of biological aging in onset and progression of disease is being actively investigated. In Parkinson’s and Alzheimer’s diseases, for example, several groups have found links between aging and neurodegeneration [206–208]. In adult-onset HD, the pathogenesis is similar; although HD patients have the gene mutation from birth, it takes several decades for the mutation to manifest; begging the question of what exactly triggers disease-onset. While it could simply be the passage of time, there is overwhelming overlap between the hallmarks of aging and cellular alteration in HD, creating the hypothesis that delaying biological aging could delay onset or progression of HD symptoms as well. In this hypothesis, the aging process plays an active role in HD pathogenesis; exacerbating the toxicity of mutant HTT (Fig. 1).
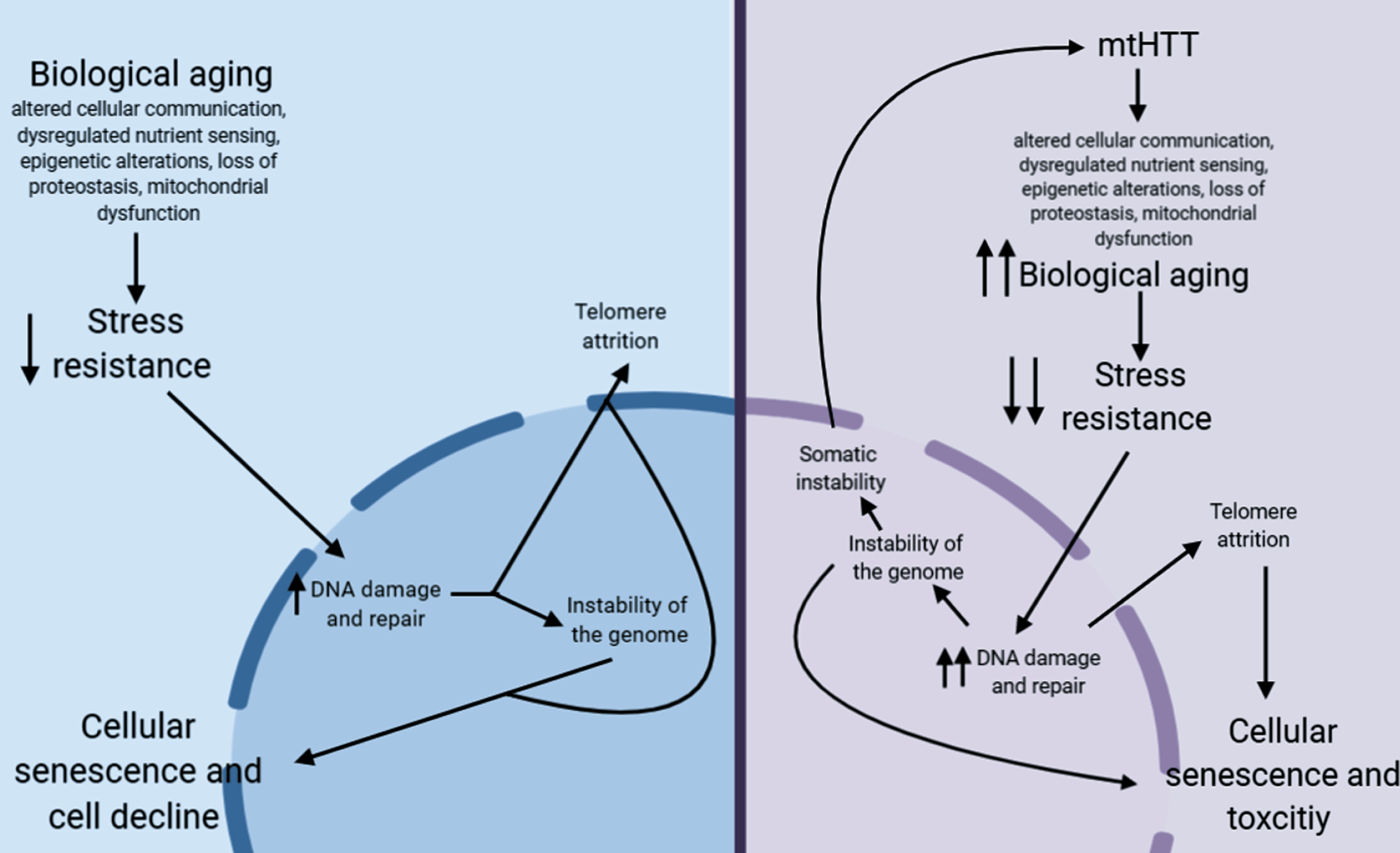
Proposed pathway for how aging may contribute to HD pathogenesis.
In non-HD neurons, biological aging occurs over time, which eventually decreases stress resistance in cells [209, 210]. This decrease in stress resistance leads to an increase in unrepaired DNA damage, which is hypothesized to cause damage to telomeres leading to telomere attrition [211, 212], and can lead to instability of the genome, particularly at microsatellite repeats [213]. Together, this leads to cellular senescence, another aging hallmark, and eventual cellular decline. In HD neurons, mtHTT accelerates aging by exacerbating several hallmarks of aging. This may lead to the hyper-susceptibility of HD neurons to stress, a further increase in DNA damage, and, as a result, accelerated telomere attrition. This increase in DNA damage could also lead to an increase in somatic instability, further increasing toxicity of mtHTT.
Another point of investigation in the role of aging in HD is how inducing aging in model systems may uncover HD-relevant phenotypes in adult-onset CAG tract length models. Currently, most models of adult-onset HD at the most common pathological repeat lengths display little to no phenotypes. Inducing aging in these models may not only uncover phenotypes, but may provide models systems that are more relevant to the pathology of adult-onset HD.
The overlap between the hallmarks of aging and cellular pathogenesis of HD as well as the accelerated aging found in HD models and patient brain provides rationale for further investigation into the role aging in HD. While we acknowledge the toxicity of mtHTT on its own, in both aging and HD, many of these ‘hallmarks’ of cellular pathogenesis converge on common pathways and can synergistically cause cellular toxicity. Thus, anti-aging therapies could be beneficial for multiple components of HD. Future work should include how modulating aging might affect pathogenesis, including somatic instability.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.