
Abstract
Background:
Cardiac ischemia/reperfusion (I/R) injury has been shown to impose deleterious effects not only on the heart but also on the brain. Our previous study demonstrated that pretreatment with a mitochondrial fusion promoter (M1) provided central neuroprotective effects following cardiac I/R injury.
Objective:
To investigate the effects of M1 given during the ischemic phase and M1 given at the beginning of reperfusion on brain pathologies following cardiac I/R.
Methods:
Male Wistar rats were randomly divided into either a sham operation (n = 6) or cardiac I/R injury (n = 18) group. Rats with cardiac I/R injury were then randomly divided into 3 subgroups: 1) Control, 2) M1 treatment during cardiac ischemia (2 mg/kg, intravenous (i.v.)), and 3) M1 treatment at the beginning of reperfusion (2 mg/kg, i.v.). After euthanasia, the brain of each rat was removed for further analysis.
Results:
Cardiac I/R injury caused brain mitochondrial dynamic imbalance, brain mitochondrial dysfunction, brain apoptosis, microglial dysmorphology, brain inflammation, tau hyperphosphorylation, and synaptic dysplasticity. M1 treatment at both time points effectively improved these parameters. M1 given during the ischemic phase had greater efficacy with regard to preventing brain mitochondrial dysfunction and suppressing brain inflammation, when compared to M1 given at the beginning of reperfusion.
Conclusions:
Our findings suggest that treatment with this mitochondrial fusion promoter prevents mitochondrial dynamic imbalance in the brain of rats with cardiac I/R injury, thereby attenuating brain pathologies. Interestingly, giving the mitochondrial fusion promoter during the ischemic phase exerted greater neuroprotection than if given at the beginning of reperfusion.
Keywords
INTRODUCTION
Cardiovascular disease remains the leading cause of mortality worldwide [1, 2], with acute myocardial infarction (AMI) being the most prevalent cause [1, 2]. Revascularization is the standard treatment for AMI, but it has the potential to lead to cardiac ischemia/reperfusion (I/R) injury, which damages both the heart and the brain [3]. Brain mitochondrial function and dynamics, blood-brain barrier (BBB) disruption, inflammation, amyloid-β plaque formation, tau hyperphosphorylation, and apoptosis are all affected by cardiac I/R injury, as shown by our previous studies [3–6].
Mitochondria, which are critical for cellular function [7–9], are highly dynamic and are maintained in a certain balance by fission and fusion processes [10, 11]. Findings from our and other studies have demonstrated that the imbalance in mitochondrial dynamics in the brain caused by cardiac I/R injury leads to apoptosis in the brain [3–5, 12–18]. Our previous research has shown that administration of a mitochondrial fusion promoter (M1) before cardiac I/R injury could have beneficial effects on the brain, including attenuation of mitochondrial dysfunction, amelioration of mitochondrial imbalance, amelioration of BBB dysfunction, reduction of activated microglia, suppression of inflammation, reduction of amyloid-β plaque formation and tau hyperphosphorylation, and reduction of apoptosis [19]. However, administration of M1 before the cardiac ischemic phase is not practical in the clinical or at-home setting.
Therefore, we investigated whether administration of M1 after cardiac ischemia onset could be viable as a neuroprotective agent to explore the importance of temporal proximity to the indexed ischemic event. We hypothesized that administration of M1 after cardiac ischemia might have beneficial effects on the brain by improving mitochondrial dysfunction, improving mitochondrial dynamic balance, improving BBB dysfunction, reducing activated microglia, suppressing inflammation, reducing tau hyperphosphorylation, and reducing apoptosis in rats with cardiac I/R injury. We also compared the effects of M1 administration during the ischemic phase and at the beginning of reperfusion on these brain parameters in rats with cardiac I/R injury.
MATERIALS AND METHODS
Experimental protocol
The experimental protocol utilized in this study was sanctioned by the Institutional Animal Care and Use Committee of the Faculty of Medicine at Chiang Mai University, Thailand. The study adhered to the ARRIVE guidelines and the approval number 05/2560 was given. Male Wistar rats weighing between 250–300 g were procured from Nomura Siam Company located in Bangkok, Thailand. All rats were housed in pairs under regulated climatic conditions of 25±0.5°C and maintained under a 12-h light/dark cycle. Rats were fed standard laboratory chow (Mouse Feed Food No. 082, C.P. Company, Bangkok, Thailand) and had unrestricted access to reverse osmosis water.
After a 1-week period of acclimatization, rats underwent either sham surgery (n = 6) or I/R surgery (n = 18), which involved 30 minutes of cardiac ischemia followed by 120 min of reperfusion, as previously reported [14]. In the I/R group, rats were randomly segregated into three subgroups: 1) control, 2) I/R surgery with M1 administration during the cardiac ischemic phase (2 mg/kg, intravenous (i.v.) injection, 15 min after ligation of the left anterior descending (LAD) coronary artery), and 3) I/R surgery with M1 administration at the start of reperfusion (2 mg/kg, i.v.). After the completion of the I/R protocols, all rats were euthanized using an overdose of Zoletil (100 mg/kg, Virbac Laboratories, Carros, France) until 1 min after breathing stopped, followed by the decapitation, and their brain tissues were rapidly harvested for the determination of brain mitochondrial isolation and function. Western blot analysis was conducted to determine the expression of brain mitochondrial dynamic proteins, brain inflammation, tau phosphorylation, synaptic plasticity, and brain apoptosis, as illustrated in Fig. 1.
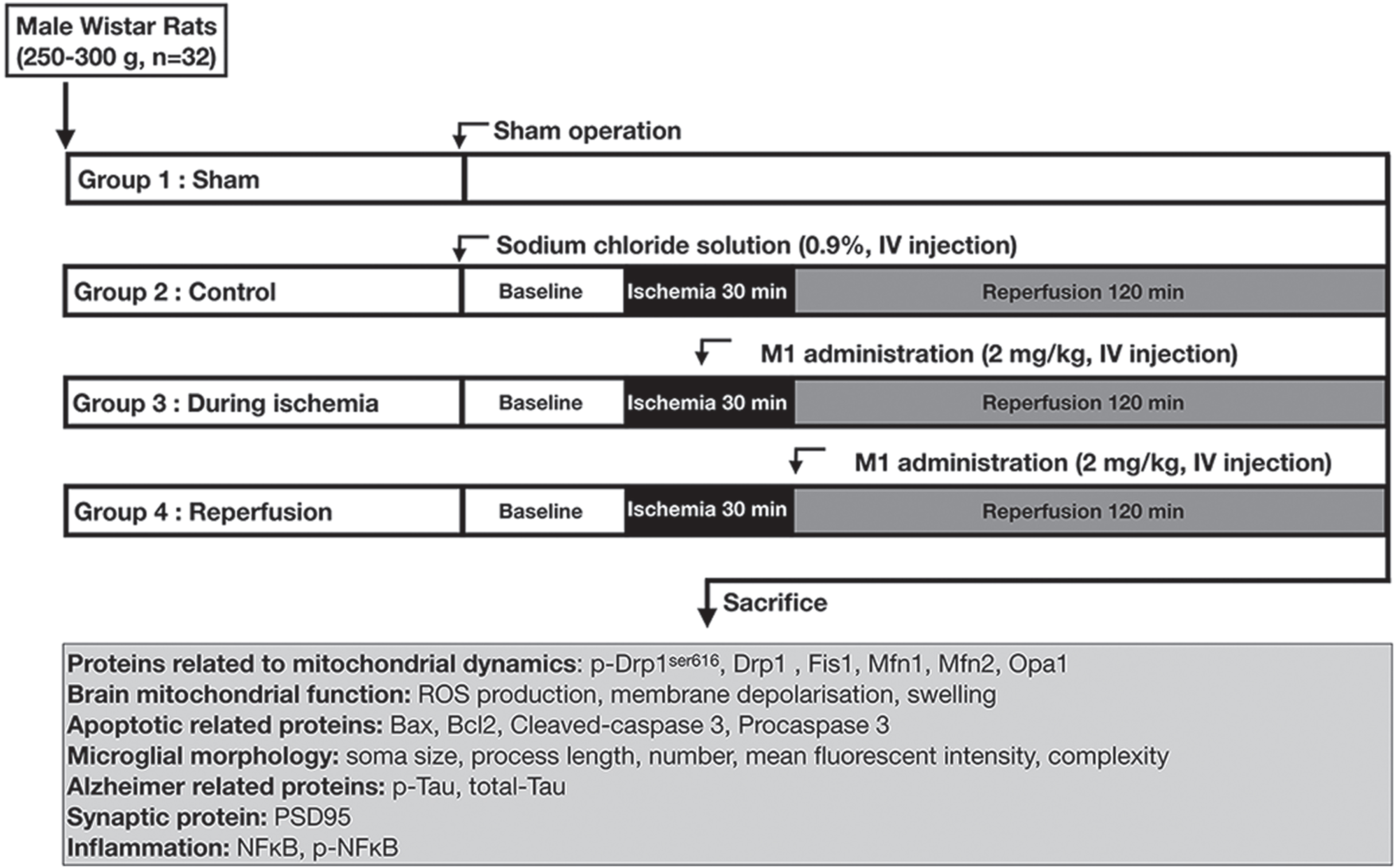
Experimental protocol of the study.
Drugs preparation
The mitochondrial fusion promoter powder (Sigma-Aldrich, MO, USA) 25 mg was dissolved in 2.5 ml of 10% Dimethyl sulfoxide (DMSO, ACI Labscan, Bankok, Thailand) for the stock solution. Then, the stock solution was diluted to 2 mg/ml by adding 10 ml of 0.9% sodium chloride solution (NSS) as described previously [19]. The dosage of the fusion promoter was calculated based on the body weight of the rats at the dosage of 2 mg/kg and the final volume was 1 ml [19]. The resulting solution was manually injected through the femoral vein. In the control group, 1 ml of NSS was injected through the femoral vein.
Surgical preparation of the cardiac I/R model
To anesthetize the rats, a combination of Xylazine (0.15 mg/kg, LBS Laboratories, Bangkok, Thailand) and Zoletil (50 mg/kg, Virbac Laboratories, Carros, France) was administered by intramuscular injection. Following anesthesia, a ventral midline incision was made, and a tracheostomy was performed. Rats were then ventilated with room air using a positive pressure ventilator (CWE, Inc., Ardmore, PA, USA). To induce cardiac ischemia, the LAD coronary artery was located and ligated by a non-absorbable silk suture 5-0 with an atraumatic 3/8 circle 12 mm reverse cut needle (Ethicon Suture Laboratories, Cincinnati, OH, USA) approximately 2 mm from its origin. The electrocardiogram and the color changes of the cardiac tissue in the ischemic region were monitored to confirm the onset of ischemia. After 30 minutes of ischemia, the ligation was released, and the ischemic myocardium was reperfused for 120 min[19].
Brain mitochondrial isolation and determination of mitochondrial function
After euthanasia, the rat brains were quickly removed and placed in a cold solution. After brain homogenization, the mitochondria of the brain were isolated by the differential centrifugation method as described previously [20]. The amount of isolated brain mitochondrial protein was determined using a bicinchoninic acid (BCA) assay kit [21].
To measure the levels of reactive oxygen species (ROS), brain mitochondrial potential (ΔΨ), and brain mitochondrial swelling, a fluorescent microplate reader was employed. For ROS measurement, brain mitochondria were incubated with dichloro-hydrofluoresceinacetate (DCFH-DA) dye, and the rise in fluorescence intensity indicated an increase in ROS production. To determine the mitochondrial potential, 5,5-,6,6-tetrachloro-1,1-,3,3-tetraethyl benzimidazolcarbocyanine iodide (JC-1) dye was used. A decrease in the ratio of red to green indicated a decrease in mitochondrial membrane potential. Additionally, changes in the absorbance of the suspension at 540 nm were measured to determine mitochondrial swelling. A decrease in the absorbance value indicated increased swelling of the mitochondria [21].
Determination of microglia morphology and images analysis
The brain tissue samples were first fixed in 4% paraformaldehyde for 24 h and subsequently immersed in 30% sucrose for 72 h. The samples were then sliced into 20μm-thick sections using a cryostat (Leica CM1950, Leica Biosystem Nussloch GmbH, Nussloch, Germany). The sections were treated with 3% peroxide to quench endogenous peroxidase activity, followed by permeabilization with 0.1% TritonX-100, blocked with 5% bovine serum albumin (BSA), and incubated with primary antibodies targeting ionized calcium-binding adapter molecule 1 (Iba-1) (ab5076, Abcam, Cambridge, MA) at 4°C, as previously described [22]. Following 24 h of incubation, the sections were washed three times with phosphate-buffered saline (PBS) and then incubated with Iba1-AlexaFluor 488 anti-goat for 1 h at 25°C. The sections were mounted using Fluoromount (Sigma-Aldrich Chemie, Steinheim, Germany), and the microglia images were captured using confocal microscopy (Olympus Fluoview FV3000, Tokyo, Japan). For morphological analysis, a three-dimensional (3D) model of microglia was reconstructed from Z-stack images using Imaris software (Bitplane, Oxford Instrument Company, AG, Zurich, Switzerland). The complexity of the microglial branching was quantified using Sholl analysis, which has been previously described [22].
Western blot analysis
To extract proteins from the brain tissue, the tissue was homogenized, centrifuged, and mixed with loading buffer. The mixture was then subjected to electrophoresis in a 10% gradient SDS-polyacrylamide gel, and the proteins were transferred to a nitrocellulose membrane. After the membranes were blocked in 5% skim milk for one hour, they were treated over night with primary antibodies, including rabbit anti-p-Drp1ser616, Dynamin related protein 1 (DRP1), (MFN1), Optic atrophy 1 (OPA1), Bcl-2-associated X protein (Bax), B-cell lymphoma 2 (Bcl-2), Procaspase-3, Kappa-light-chain-enhancer of activated B cells (NF-κB), p-NF-κB, p-TauThr181, Tau, and Postsynaptic density protein 95 (PSD95). After treatment with a secondary goat anti-rabbit antibody coupled with horseradish peroxidase, protein bands were identified using enhanced chemiluminescent (ECL) western blot detection reagents and the ChemiDoc Touch Imaging System. Anti-β-actin was used as a loading control. The average signal intensity (arbitrary units) of the bands was measured using Image J software [3–5, 19].
Statistical analysis
All data in the study are presented as mean±S.E.M. To compare differences between groups, a one-way ANOVA followed by LSD post hoc test was used. A p < 0.05 was considered statistically significant.
RESULTS
The M1 treatment during the ischemic phase or at the beginning of reperfusion equally attenuated brain mitochondrial dynamic imbalance following cardiac I/R injury
The results demonstrate that cardiac I/R injury disrupts the balance of mitochondrial dynamics in the brain. Specifically, rats in the control group exhibited a reduction in brain mitochondrial fusion, as evidenced by decreased expression of MFN1 and OPA (Fig. 2A, B, D). However, cardiac I/R injury did not result in alterations in brain mitochondrial fission, as evidenced by the absence of changes in the p-DRP1ser616/DRP1 ratio across all groups (Fig. 2C, D). Intriguingly, treatment with M1 during the ischemic phase and at the beginning of reperfusion was found to mitigate the imbalance in brain mitochondrial dynamics induced by cardiac I/R injury, as demonstrated by an increase in MFN1 and OPA expression (Fig. 2A, B, D). These findings suggest that cardiac I/R injury leads to an imbalance in brain mitochondrial dynamics, which can be effectively ameliorated by treatment with M1 at either the ischemic phase or the beginning of reperfusion.
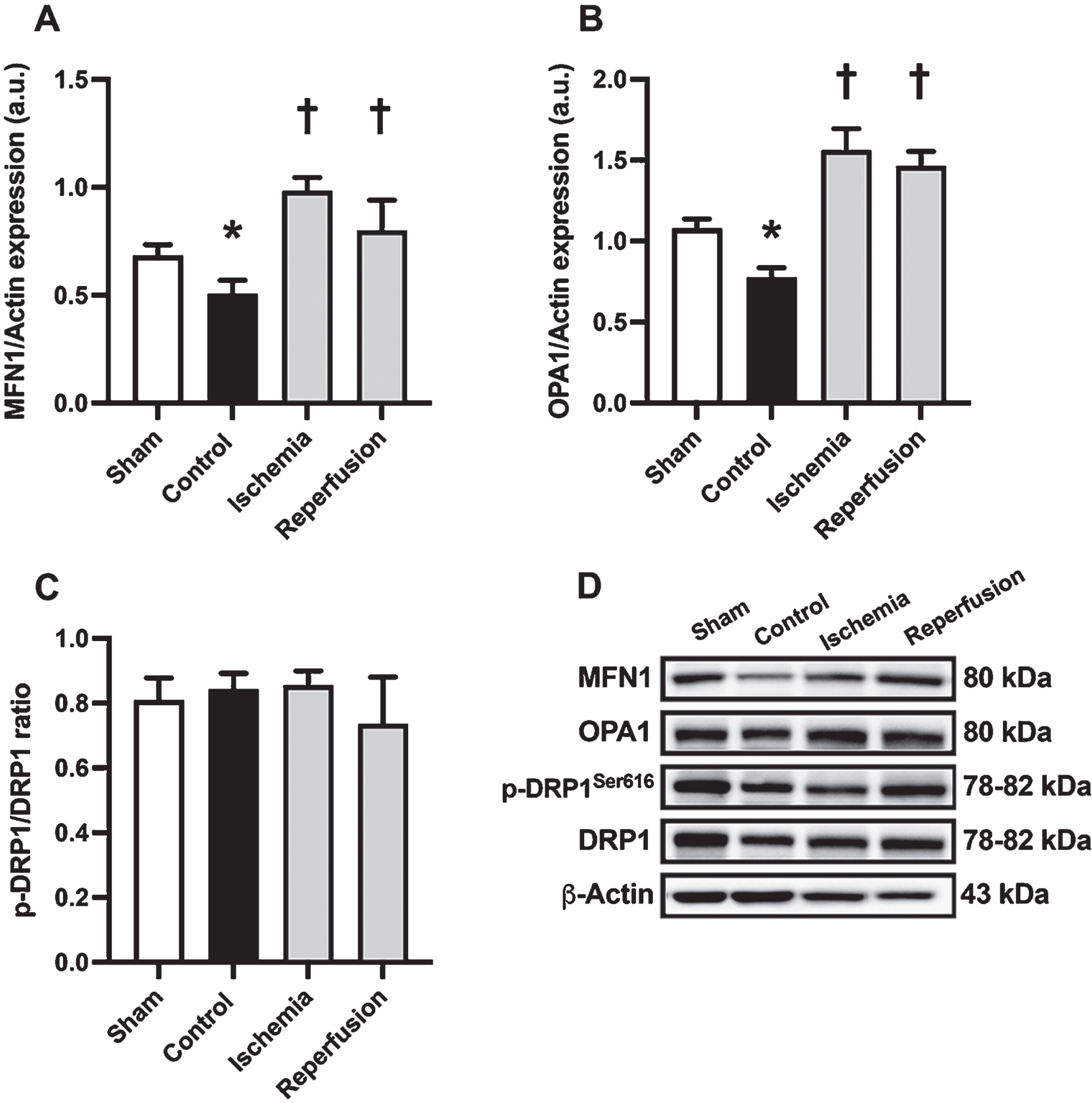
The effects of M1 given during the ischemic phase or at the beginning of reperfusion on brain mitochondrial dynamics balance in rats with cardiac I/R injury. A, B) The expression of mitochondrial fusion related proteins, including MFN1 and OPA1. C) The expression of mitochondrial fission proteins, including the p-DRP1/DRP1 ratio. D) Representative western blot images showing protein bands. n = 6/group; *p < 0.05 versus Sham; †p < 0.05 versus Control. DRP1, dynamin related protein 1; MFN1, mitofusin 1; OPA1: optic atrophy 1.
The M1 treatment during the ischemic phase had greater benefit with regard to brain mitochondrial function than M1 given at the beginning of reperfusion
In order to gain further insight into the effects of M1 administered during the ischemic phase and at the beginning of reperfusion on brain mitochondrial function, we measured ROS production, mitochondrial membrane potential, and the swelling of isolated brain mitochondria (Fig. 3). Our results demonstrated that cardiac I/R injury significantly increased ROS production, mitochondrial depolarization in isolated brain mitochondria, and decreased absorbance at 540 nm (Fig. 3A-C). Treatment with M1 during the ischemic phase was found to improve brain mitochondrial function, as evidenced by a decrease in brain ROS production, decreased brain mitochondrial depolarization, and increased absorbance value (Fig. 3A-C). However, M1 administered at the beginning of reperfusion was only able to effectively reduce brain ROS production while not having any significant impact on brain mitochondrial depolarization or brain mitochondrial swelling (Fig. 3A-C). These findings suggest that M1 administered during the ischemic phase may confer greater benefits to brain mitochondrial function than being given at the beginning of reperfusion.
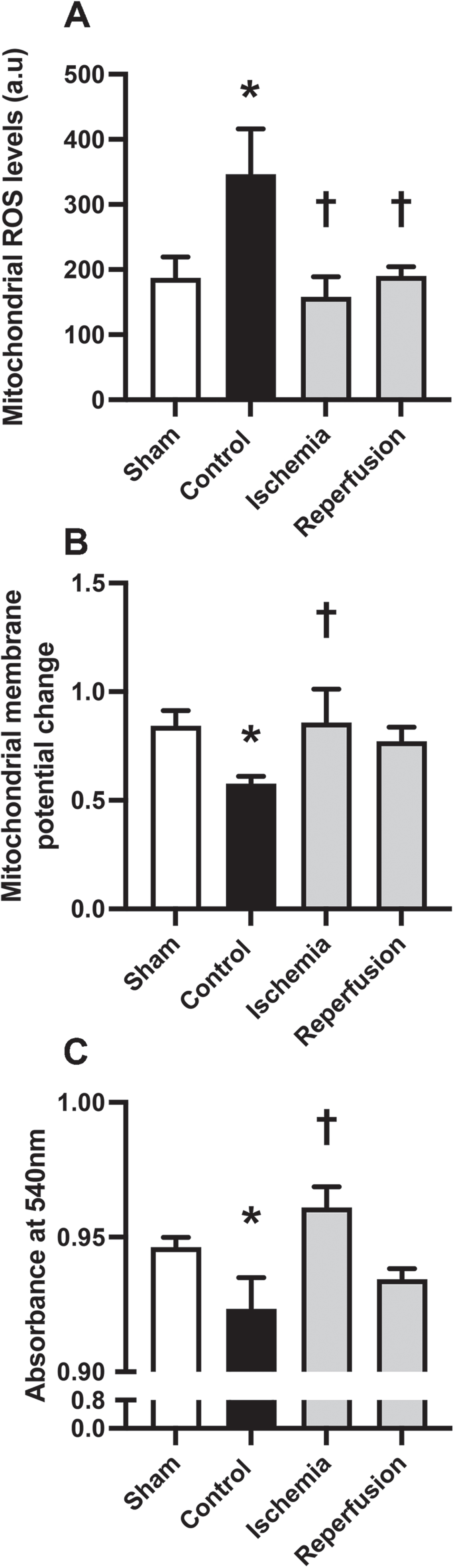
The effects of M1 given during the ischemic phase or at the beginning of reperfusion on brain mitochondrial function in rats with cardiac I/R injury. A) Brain mitochondrial ROS production. B) Brain mitochondrial membrane potential as indicated by a red per green fluorescence intensity ratio. C) Brain mitochondrial swelling as indicated by absorbance value. n = 6/group; *p < 0.05 versus Sham; †p < 0.05 versus Control. ROS, reactive oxygen species.
M1 given during the ischemic phase or given at the beginning of reperfusion decreased brain apoptosis to an equal extent following cardiac I/R injury
To investigate the potential effects of cardiac I/R injury on brain apoptosis, we measured the Bax/Bal2 ratio and the cleaved-caspase 3/procaspase 3 ratio (Fig. 4). Our results indicated that rats in the control group showed a significantly increased Bax/Bal2 and cleaved-caspase 3/procaspase 3 ratios (Fig. 4A, B). Intriguingly, treatment with M1 during the ischemic phase or at the beginning of reperfusion was found to reduce brain apoptosis equally, as demonstrated by a decrease in both the Bax/Bal2 ratio and the cleaved-caspase 3/procaspase 3 ratio (Fig. 4A, B). These findings suggest that M1 administered during the ischemic phase or at the beginning of reperfusion effectively reduced brain apoptosis in rats with cardiac I/R injury.
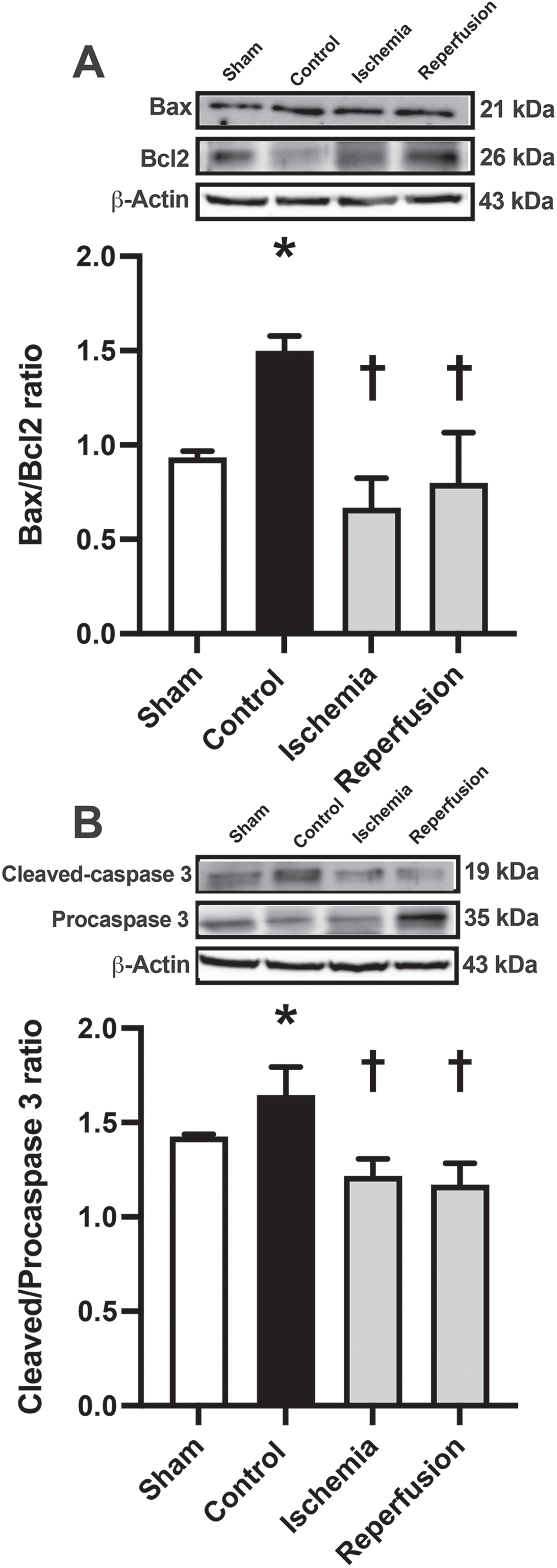
The effects of M1 given during the ischemic phase or at the beginning of reperfusion on brain apoptosis in rats with cardiac I/R injury. A) The Bax/Bal2 ratio. B) The cleaved-caspase 3/ Procaspase 3 ratio. n = 6/group; *p < 0.05 versus Sham; †p < 0.05 versus Control. Bal2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein.
M1 given during the ischemic phase and given at the beginning of reperfusion preserved microglial morphology following cardiac I/R injury
We conducted further investigations in order to examine the potential effects of M1 administration during the ischemic phase and at the beginning of reperfusion on microglial morphology in rats with cardiac I/R injury (Fig. 5A). Our results demonstrated a significant increase in microglial number in the control group, which was effectively reduced in M1-treated rats with cardiac I/R injury at both time points (Fig. 5B). However, we observed no significant difference in the mean immunofluorescence intensity of Iba-1 positive cells across all groups (Fig. 5C). Additionally, rats in the control group exhibited a decrease in both the volume and length of microglial processes (Fig. 5D, E). However, M1 administration during the ischemic phase significantly increased both the volume and length of microglial processes, whereas M1 administration at the beginning of reperfusion increased only the volume of the microglial processes without any changes in their length (Fig. 5D, E). The rats in the control group also exhibited a decrease in segments, branch points, and end points of microglia, which were effectively increased in M1-treated rats at both time points (Fig. 5F-H). We also conducted a Sholl analysis to investigate microglial complexity (Fig. 5I, J). Our results showed a significant decrease in the area under the curve of Sholl intersections in the control group, which was effectively increased in M1-treated rats at both time points (Fig. 5I, J). These findings suggest that M1 administration during the ischemic phase or at the beginning of reperfusion effectively preserved microglial morphology in rats with cardiac I/R injury.
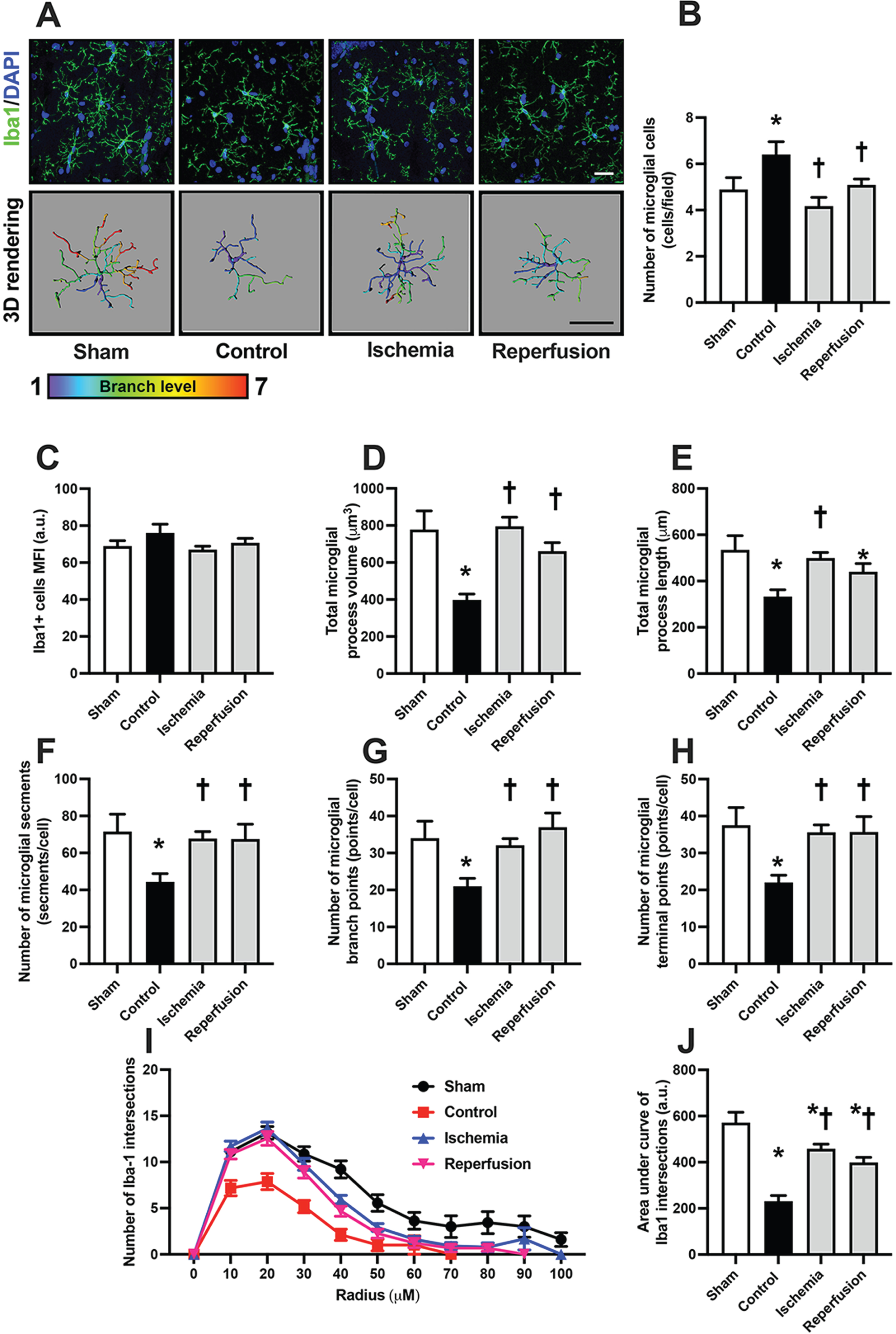
The effects of M1 given during the ischemic phase or at the beginning of reperfusion on microglial morphology in rats with cardiac I/R injury. A) Representative images of microglial morphology under confocal microscopy at the CA1 region of the hippocampus by immunofluorescent staining of DAPI (blue), Iba1 (green), colocalization (Iba1/DAPI) (bar = 50μm), and the 3D structure of microglial morphology by Imaris software (bar = 50μm). B) The number of Iba-1 positive cells. C) Mean immunofluorescent intensity of Iba-1 positive cells. D) Total process volume of Iba-1 positive cells. E) Total process length of Iba-1 positive cells. F) Mean segment number of Iba-1 positive cells. G) Mean branch points of Iba-1 positive cells. (H) Mean end points of Iba-1 positive cells. I, J) Microglial ramification as indicated by Sholl analysis and area under the curve of Sholl intersections. n = 6/group; *p < 0.05 versus Sham; † p < 0.05 versus Control. 3D, three dimensional; Iba-1, ionized calcium-binding adapter molecule 1
M1 given during the ischemic phase or given at the beginning of reperfusion attenuated brain inflammation, decreased tau hyperphosphorylation, and improved synaptic plasticity following cardiac I/R injury
In this study, we observed that rats with cardiac I/R injury showed increased brain inflammation, as evidenced by an elevation in the p-NFκB/NFκB ratio, which was significantly decreased by M1 treatment during the ischemic phase but not at the beginning of reperfusion (Fig. 6A, D). Notably, the control group exhibited an elevated p-Tau/Tau ratio, indicating increased tau hyperphosphorylation, whereas M1 treatment at both time points led to a decrease in this ratio (Fig. 6B, D). Additionally, we found that the expression of PSD95, a synaptic protein, was significantly decreased in the control group, while M1 treatment at both time points increased its expression to an equal extent (Fig. 6C, D). These results suggest that M1 given during the ischemic phase or at the beginning of reperfusion can effectively attenuate brain inflammation, decrease tau hyperphosphorylation, and improve synaptic plasticity following cardiac I/R injury.
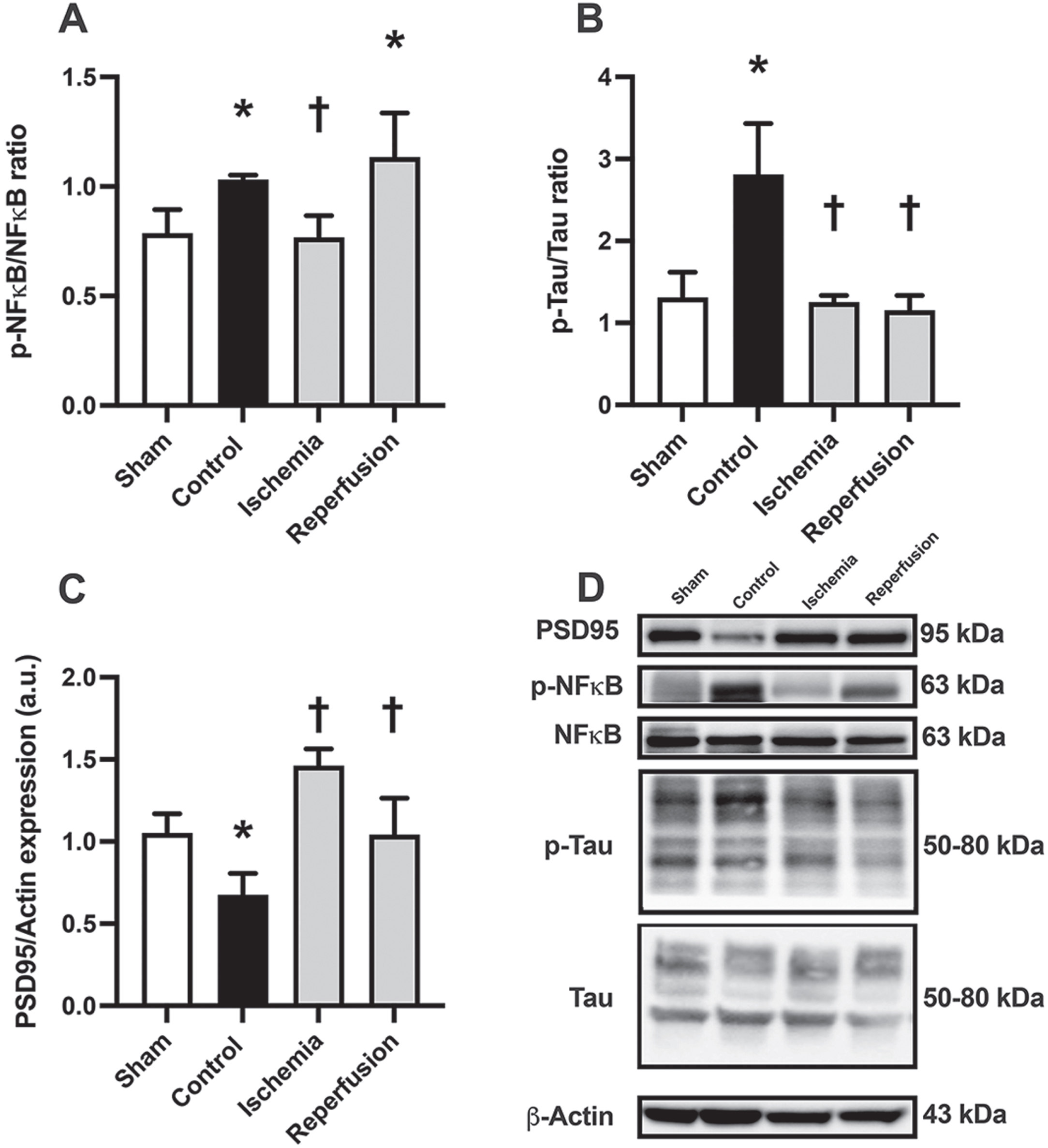
The effects of M1 given during the ischemic phase or at the beginning of reperfusion on brain inflammation, Alzheimer’s disease related proteins, and synaptic proteins in rats with cardiac I/R injury. A) Brain inflammation as indicated by p-NF-κB/NF-κB ratio. B) The expression of Alzheimer’s disease related proteins, including p-Tau/Tau ratio. C) The expression of synaptic proteins, including PSD95. D) Representative western blot images showing protein bands. n = 6/group; *p < 0.05 versus Sham; †p < 0.05 versus Control. NF-κB, kappa-light-chain-enhancer of activated B cells; PSD95, postsynaptic density protein 95.
DISCUSSION
There were several key findings in this study. Firstly, cardiac I/R resulted in brain mitochondrial dynamic imbalance, brain mitochondrial dysfunction, cell apoptosis, microglial dysmorphology, brain inflammation, hyperphosphorylation of Tau, and synaptic dysplasticity. Second, administration of M1 during the ischemic phase or at the beginning of reperfusion effectively attenuated the brain pathologies caused by cardiac I/R injury by improving the imbalance of mitochondrial dynamics in the brain, reducing brain mitochondrial dysfunction, decreasing brain apoptosis, suppressing inflammation in the brain, preserving microglial morphology, reducing tau hyperphosphorylation, and improving synaptic plasticity. Lastly, M1 administered during the ischemic phase was more effective in improving brain mitochondrial function and suppressing inflammation in the brain than M1 administered at the beginning of reperfusion.
AMI not only causes cardiac dysfunction but also leads to brain dysfunction and abnormalities. Studies have revealed that cardiac I/R injury triggers oxidative stress and inflammation in the bloodstream, leading to disruption of BBB tight junction proteins and hence increasing BBB permeability, which allows entry of systemic ROS and pro-inflammatory cytokines into the brain [3, 5]. This can lead to oxidative stress and inflammation in the brain. Moreover, cardiac I/R injury can result in a mitochondrial dynamic imbalance in the brain, as indicated by a reduction in mitochondrial fusion [19]. This study also found that mitochondrial fusion, but not fission, was impaired in response to cardiac I/R injury. Although the mechanism underlying the selective decline in mitochondrial fusion in the brain is unknown, it is possible that mitochondrial fusion in the brain is more susceptible to change in response to cardiac I/R injury than mitochondrial fission. As a result, an imbalance in brain mitochondrial dynamics caused by cardiac I/R injury may lead to mitochondrial dysfunction in the brain [14], which can trigger cellular apoptosis.
Brain inflammation is a key mechanism responsible for the pathologies in the brain that occur after cardiac I/R injury [3, 5]. Several studies have demonstrated that microglial function is altered under conditions of cardiac I/R [23, 24], with an increase in both the activity and number of microglia [23]. Moreover, changes in microglial morphology have been detected, indicating that these cells adopt an amoeboid form and possibly produce more proinflammatory cytokines [13, 26]. All of these events lead to an increase in brain inflammation, which subsequently affects brain mitochondrial function, causing neuronal apoptosis [3, 13]. According to a previous study, neuronal apoptosis was observed after cardiac I/R injury, which may have been related to neuronal degeneration in the hippocampus [23]. Several other studies have linked brain inflammation to an increase in proteins related to Alzheimer’s disease [12, 27–29]. Consistent with the findings in those studies, the present study revealed that tau hyperphosphorylation also increased after cardiac I/R injury, possibly due to cardiac I/R-induced brain inflammation. Synaptic plasticity also decreased under neuroinflammatory conditions, as evidenced by a reduction in synaptic proteins [25, 31], likely due to the inflammatory response to cardiac I/R injury. Overall, cardiac I/R injury induces oxidative stress in peripheral tissues and the brain, leading to microglial activation, impairment of brain mitochondrial function, disrupting brain mitochondrial dynamic balance, increasing brain apoptosis, and causing brain dysplasticity.
M1 has been identified as a powerful promoter of mitochondrial fusion [32]. Increasing evidence indicates that changes in mitochondrial dynamics are crucial, particularly in response to injury [14, 34]. By controlling the expression of Mfn1 and Mfn2 at the outer mitochondrial membrane, M1 efficiently promotes mitochondrial elongation [32]. Furthermore, the administration of M1 at a dosage of 2 mg/kg has been shown to have cardioprotective effects against cardiac I/R injury [14, 34]. Pharmacological manipulations using M1 or other mitochondrial fusion promoters have been employed in the context of extended cardioprotection to mitigate brain damage and restore the balance of mitochondrial dynamics, suggesting the potential of M1 for conferring neuroprotection [35]. One of our previous studies showed that administration of M1 before cardiac ischemia effectively improved both brain mitochondrial dynamic balance and brain mitochondrial function [19]. In addition, the present study showed that administration of M1 during the ischemic phase and at the beginning of reperfusion improved brain mitochondrial dynamic balance by upregulating the expression of Mfn1 and OPA1. While M1 treatment at both time points suppressed the production of ROS, only M1 given during the ischemic phase restored brain mitochondrial membrane potential and reduced brain mitochondrial swelling. These results may be attributed to the acute therapy because M1 was administered to the animals in a single dose. As a result, excessive calcium-induced neurotoxicity at the beginning of reperfusion may not have been counteracted by a single dose of M1. These findings suggest that the effect of M1 extends beyond the promotion of mitochondrial fusion. We have gone on to speculate that M1 may exert an antioxidant function within the cells that could be associated with an improvement in brain mitochondrial function.
In addition to oxidative stress, brain inflammation has also been suggested as a potential cause of brain damage following cardiac I/R injury [3, 5]. It has been reported that following cardiac I/R injury, peripheral macrophages and proinflammatory cytokines from the bloodstream can activate resident microglia, leading to brain inflammation [19]. This inflammation may worsen as activated microglia release more proinflammatory cytokines [36–39]. Our recent study found that pretreatment with M1 reduced macrophage infiltration and microglial activation [19]. The present study demonstrated still further that administration of M1 during the ischemic phase and at the beginning of reperfusion effectively preserved microglial morphology and reduced microglial activation, resulting in a reduction in brain inflammation. In addition, we speculated that M1 might exert an anti-inflammatory effect, which may be associated with a reduction in tau hyperphosphorylation and an increase in synaptic proteins. Although the present study did not determine the pro-inflammatory cytokine levels due to the limited brain tissue, the elevation in the p-NFκB/NFκB ratio indicated brain inflammation in rats with cardiac I/R injury, which was attenuated by M1 treatment. Consistently, our previous studies showed that M1 treatment effectively suppressed IL-6 and TNFα mRNA levels in rats with doxorubicin-induced cardiotoxicity and neurotoxicity [22, 40]. Further studies should determine the pro-inflammatory cytokine levels in the brain to determine the effect of M1 treatment on brain inflammation in rats with cardiac I/R injury.
It is worthy to note that although the present study demonstrated tau hyperphosphorylation, one of markers for Alzheimer’s disease, the pathological mechanisms between cardiac I/R injury and Alzheimer’s disease are different [5, 41–45]. Alzheimer’s disease is caused by the prolonged accumulation of amyloid-β plaque, tau hyperphosphorylation, and long-term brain inflammation [42–45], whereas cardiac I/R injury is caused by the accumulation of pro-inflammatory cytokines, brain oxidative stress, and brain mitochondrial dysfunction due to ischemia and reperfusion injury with a short period of time [5, 41]. It is plausible that cardiac I/R injury might accelerate or/and aggravate Alzheimer’s disease pathologies since we showed an elevation of tau phosphorylation. Further studies should investigate the brain pathologies in cardiac I/R injury with Alzheimer’s disease models.
Cardiac I/R injury has been linked to the induction of apoptosis, which is believed to be triggered by inflammation and mitochondrial dysfunction in the brain [46]. This study confirmed that there was an increase in both the Bax/Bcl2 ratio and the cleaved-caspase 3/procaspase3 ratio in response to cardiac I/R injury, both of which were ameliorated by M1 treatment. These findings suggest that M1, a mitochondrial fusion promoter, can mitigate brain inflammation and dysfunction, thereby reducing the vulnerability of the brain to apoptosis following cardiac I/R injury.
Conclusion
The therapeutic administration of this mitochondrial fusion promoter demonstrated notable neuroprotective effects by improving the balance of brain mitochondrial dynamics, mitigating mitochondrial dysfunction, reducing brain apoptosis, preserving microglial morphology, suppressing brain inflammation, reducing tau hyperphosphorylation, and enhancing synaptic plasticity. The beneficial effects of M1 were more pronounced when administered during the ischemic phase than at the beginning of reperfusion, indicating its potential use as a therapeutic agent to minimize brain damage after cardiac I/R injury. These findings are relevant in a clinical context, as M1 also shows promise with regard to providing protective effects for individuals after cardiac ischemia has occurred.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank Dr. Poomarin Surinkaew, Dr. Kodchanan Singhanat, and Ms. Thidarat Jaiwongkam for the laboratory technical assistance.
FUNDING
This work was supported by Thailand Science Research and Innovation-Chiang Mai University (Fundamental Fund 2565 to TC); the Research Grant for Young Researchers from National Research Council of Thailand (NRCT) and Chiang Mai University (N42A660463 to TC); the Distinguished Research Professor Grant from the National Research Council of Thailand (N42A660301 to SCC); the Chiang Mai University Center of Excellence Award (NC); and the NSTDA Research Chair grant from the National Science and Technology Development Agency Thailand (NC).
CONFLICT OF INTEREST
Siriporn C. Chattipakorn is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author upon reasonable request.