
Abstract
Severe acute respiratory disease coronavirus 2 (SARS-CoV-2) is responsible for the coronavirus disease 2019 (COVID-19) pandemic. Although a primarily respiratory disease, recent reports indicate that it also affects the central nervous system (CNS). Over 25% of COVID-19 patients report neurological symptoms such as memory loss, anosmia, hyposmia, confusion, and headaches. The neurological outcomes may be a result of viral entry into the CNS and/or resulting neuroinflammation, both of which underlie an elevated risk for Alzheimer’s disease (AD). Herein, we ask: Is COVID-19 a risk factor for AD? To answer, we identify the literature and review mechanisms by which COVID-19-mediated neuroinflammation can contribute to the development of AD, evaluate the effects of acute versus chronic phases of infection, and lastly, discuss potential therapeutics to address the rising rates of COVID-19 neurological sequelae.
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) emerged in late 2019 as the highly transmissible and pathogenic coronavirus disease 2019 (COVID-19), resulting in a broad spectrum of symptoms ranging from fevers to various organ dysfunctions [1]. Although initially regarded as primarily a respiratory disease, emerging data suggest that SARS-CoV-2 carries the capacity to invade the central nervous system (CNS), causing various neurological problems including confusion, headache, anosmia (loss of smell), ageusia (loss of taste), cerebrovascular disease, encephalitis, and demyelinating pathologies [2–4]. SARS-CoV-2 has an elevated propensity to damage the CNS through aberrant activation of immune responses leading directly and indirectly to neurotoxicity [5]. Recent literature has also identified that the proteome of SARS-CoV-2 self-assembles into neurotoxic amyloids, suggesting that these aggregates induce the onset of neurological symptoms following infection [6]. Moreover, during infection, there is augmented risk of chronic brain damage through cerebral hemorrhage, endothelial and blood-brain barrier (BBB) impairments [7, 8], and increased levels of pro-inflammatory cytokine within the CNS [9, 10]. In COVID-19 cases with severe non-neurologic systemic infection and inflammation, subsequent long-term decline in cognition and neurodegeneration, and increased atrophy of the hippocampus were observed; for example, COVID-19 patients with acute respiratory distress syndrome (ARDS) had increased levels of cognitive decline and neurodegeneration [11]. In cases of COVID-19 with clinical manifestations indicating CNS involvement, initial reports from Wuhan, China reported that almost 25% of hospitalized patients experienced headaches, confusion, and disorientation— all of which reflect non-specific encephalopathy [12]. Emerging data have also found neurological lesions in brains of COVID-19 patients, although it is not known whether this is due to direct viral action or from the indirect effects of hypoxia, cytokine storm, or cerebrovascular injury [13–15]. Moreover, a number of growing case reports and clinical observations suggest neurological disturbances following COVID-19 infection [16, 17].
According to the World Health Organization (WHO), there have been over 500 million global cases of confirmed COVID-19 [18]. Undoubtedly, that number may be far greater. The emerging COVID-19 clinical observations combined with the knowledge that chronic inflammation is now recognized as a risk factor for Alzheimer’s disease (AD), raises the important question: Is COVID-19 a risk factor for AD? Given the worldwide incidence and prevalence of COVID-19, combined with the devastating personal health and socioeconomic impact of AD, this is a connection which would have immense public health consequences on a global scale. From the 500 million confirmed COVID-19 cases, even if 1% of those individuals go on to develop AD, that adds an additional 5 million cases to the already growing global burden of AD. In 2020, there were 50 million people living with dementia. Furthermore, considering that the rates of dementia are estimated to double every 20 years, COVID-19 may lead to an immediate increase of up to 10% of global cases.
In this review, we discuss the mechanisms involved in COVID-19-mediated neuroinflammation, the relative risk of AD and cognitive decline following infection, focussing primarily on the associated pathological mechanisms and biomarkers.
ACUTE VERSUS LONG COVID-19
Although initially a respiratory disease, a large volume of evidence now suggests that COVID-19 is a multi-organ disease that can manifest into a wide range of pathologies [19], lasting weeks beyond recovery. Recent studies report an increasing number of patients with extended unresolved symptoms and abnormalities following the acute phase of COVID-19 infection [20]. One study found that almost 30% of COVID-19 patients reported persistent and ongoing symptoms lasting for up to 9 months post-infection [21]. While acute COVID-19 is generally defined as lasting 4 weeks from symptomatic onset, long or post-acute sequalae of COVID-19 (PASC) diagnoses include ongoing persistent symptoms and complications beyond the initial 4 weeks [22]. Long COVID can be further classified into 2 subcategories: subacute/ongoing COVID-19 that describes symptoms observed for up to 8 weeks post-acute infection; and chronic/post-COVID-19 which includes symptoms persisting beyond 12 weeks from the onset of infection and which is not explained by separate diagnoses [20] (Fig. 1).
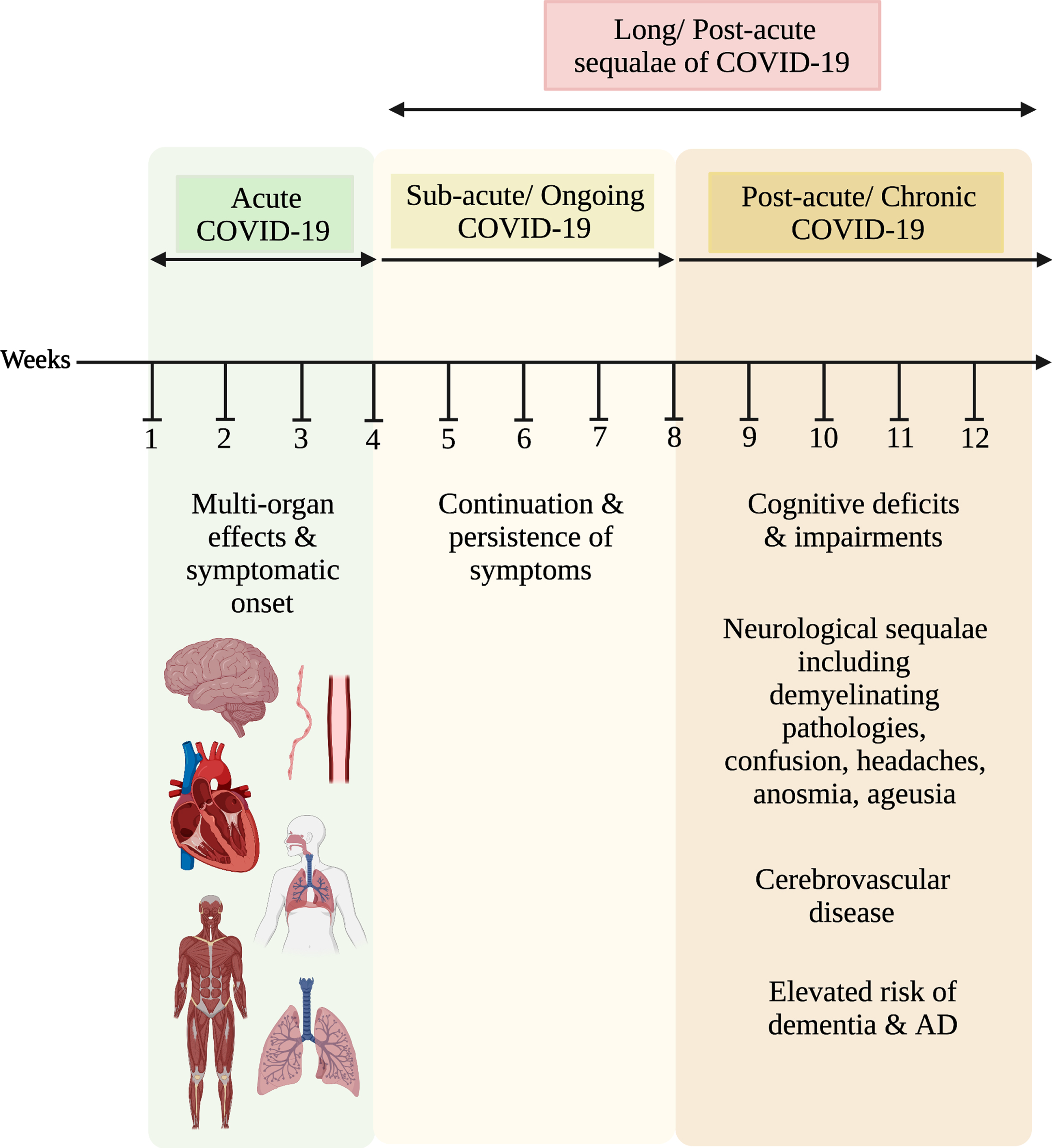
Timeline of COVID-19 infection and associated symptoms. The acute phase describes the average course of infection from onset of symptoms to the point at which replication-competent SARS-CoV-2 is not detected. At this stage, patients generally experience multi-organ effects and symptoms of hyposmia, ageusia, respiratory complications leading to severe cough, shortness of breath (from either respiratory or cardiovascular dysfunction), chest pain, arthralgia, myalgia, muscle weakness [23], and alterations in blood pressure [24]. Next is the long or postacute sequelae of COVID-19 which underlie the persistence and exacerbation of initial symptoms. Long COVID-19 is then divided into two categories: 1) sub-acute or ongoing COVID-19 stage (from weeks 4-8) which involves the persistence of symptoms observed in the acute stage; 2) Post-acute or chronic COVID-19 which involves cognitive deficits and impairments, neurological sequalae, and cerebrovascular pathologies. These persistent and worsened symptoms often elevate the risk of dementia, and ultimately lead to AD.
Not all acute COVID-19 patients develop PASC. Acute cases may vary in terms of both severity and length; some cases may appear asymptomatic, others require hospitalization and/or mechanical ventilation [25], lasting on average up to 4 weeks [26]. However, those that have prolonged acute infection then go on to develop chronic and persistent symptoms [26–28] which underlie long COVID, post-acute COVID-19 syndrome, or PASC [22].
Based on the global case counts of COVID-19, over 1.1% of confirmed positive patients have died. This leaves approximately 98.9% of COVID-19 survivors [18], many of whom have reported numerous neurological [29] and cognitive sequalae post-infection [30]. Cognitive symptoms are mostly reported in cases of acute [12] and subacute stages [31] of COVID-19. As previous coronavirus (SARS and MERS) infections have also demonstrated similar reports of ongoing symptoms [32, 33], it is not surprising that a growing body of evidence is emerging on the acute and chronic effects of COVID-19. Furthermore, as residual symptoms of COVID-19 were generally found to improve over time, neurological symptoms have been reported to last longer [34] and in some cases, develop into secondary illnesses that catalyze the development of AD [35–37]. Evidence for this comes from a study in which 7 months following infection, 88% of patients experienced cognitive dysfunction and memory deficits, suggesting that even by then, neurological symptoms have not resolved [27]. In addition, the global incidence rate of neurological complications linked to COVID-19 from both the acute and subacute states ranges from 35–85% [12, 38–40]. This leaves a major question begging to be answered: is acute or long COVID-19 a risk factor for AD? Perhaps it may be a culmination of all stages of infection; or, initial acute phase driving the start of far-reaching inflammatory responses; or, subacute/ongoing COVID-19 as a chronic immune response to the residual effects of SARS-CoV-2 [20]; or, chronic/post-COVID-19 underlying the persistent symptoms affecting brain function and cognition [26].
In a study evaluating COVID-19 patients in the sub-acute phase, Hosp et al. identified impaired neocortical function and cognitive decline in a large proportion of patients [31], suggesting that the risk of dementia is prevalent up to 4 weeks post-infection. In a recent longitudinal cohort study, almost 64% of patients reported neurological deficits without improvements lasting a year following acute infection [41]. Furthermore, a large cohort study by Søraas et al. found that that 8 months following a mild case of COVID-19 confirmed by a positive SARS-CoV-2 test, patients reported increased memory problems, indicative of worsened health and progression of PASC [42]. These findings contribute to the hypothesis that even in relatively mild illness at onset, PASC remains as a contributor to the onset of neurological and neurocognitive complications, ultimately increasing the risk of AD. Despite the fact that almost 80% of SARS-CoV-2 infected patients have little to no respiratory manifestations, and do not require hospitalization for pneumonia or hypoxemia [43, 44], a large proportion of data on cognitive complications derives from critically ill and/or hospitalized COVID-19 patients [45]. Yet, patients with mild cases of COVID-19 report persistent and exhausting neurological manifestations affecting cognition and memory, as well as brain fog, headaches, dysgeusia, anosmia, and fatigue [46]. However, like PASC from mild cases, survivors of severe COVID-19 also report cognitive deficits, with 1-year follow-up studies indicating similar cognitive test results as those diagnosed with moderate traumatic brain injury [45]. Although cognitive impairments are not absolutely correlated to the development of AD, a long-term study over 4 years found a 27% increased risk of mild cognitive impairment and a 14% elevated risk of dementia in older populations with subjective memory loss [47]. Moreover, while studies on the different stages of COVID-19 infection provide insight on the prevalence of neurocognitive sequalae, it leaves our question unresolved. It is thus probable that chronic COVID-19 is relatively independent from the severity of illness and is more likely explained by the molecular mechanisms that once initiated during the course of illness continue to progress to the post-acute/chronic state.
COVID-MEDIATED NEUROINFLAMMATION
SARS-CoV-2 enters the host to infect numerous cell types including macrophages, neutrophils, and dendritic cells [48]. The first immune response upon infection is the production of type 1 interferons (IFNs) [49]. IFNs communicate downstream immune responses depending on the detection of pathogen or damage associate molecular patterns (PAMPS/DAMPS) [50]. In the IFN response to SARS-CoV-2, production of CD8+ cytotoxic T cells and CD4+ helper T cells mediate downstream B-cell responses leading to IgM and IgG antibody production [48]. In SARS-CoV-2-infected cells, activation of T cells, B cells, natural killer cells, and macrophages augment the production cascade of pro-inflammatory cytokines and chemokines, referred to as hypercytokinemia or a “cytokine storm” [51]. A number of cytokines are involved in this phenomenon including tumor necrosis factor (TNF), IL-6, IL-10, and colony-stimulating factors [52]. Due to the nature of the cytokine storm, interactions of innate and adaptive immune systems lead to a pathogenic and hyperactive immune response, which can ultimately be fatal [52].
The blood-brain barrier (BBB) consists of endothelial cells that bar passage of pathogens like viruses from the systemic circulation into the CNS. The BBB also maintains CNS homeostasis by inhibiting neurotoxic agents from penetrating into the brain [7]. During COVID-19 infection, viral entry into the brain has been reported to be via the olfactory mucosa [53], vasculature [54], brainstem [55], or through neuroinvasive mechanisms [56]. Interactions of mural cells, immune cells, glial cells, and neural cells in the neurovascular unit, mediated by the paracellular endothelial tight junctions, maintain the barrier properties of the BBB [57]. In the case of BBB disruption or breakdown, this leads to entry of microbials from the systemic circulation into the brain to induce inflammatory immune responses (possibly initiating the neurodegenerative pathways involved in AD) [57–59]. Additionally, BBB breakdown is associated with neurovascular dysfunction and impaired cerebral blood flow [57, 60]. Ensuring the integrity of the BBB is therefore critical for maintaining control of brain interstitial fluid responsible for synaptic function and neuronal connectivity [57].
At the molecular level, SARS-CoV-2 enters the CNS through binding to the angiotensin converting enzyme 2 (ACE2) receptor on BBB endothelial cells and infects macrophages and monocytes of the CNS [61]. These events lead to BBB protective dysfunction, thereby contributing to neural inflammation via entry of plasma factors into the brain that activate glial cells and induce immune cell infiltration [62]. Microglial cells, once active, take on the pro-inflammatory M1 phenotype [63]. Recent data suggest that the associated neurological symptoms of COVID-19 may be a result of chronic changes in the neurons, glial cells and brain vasculature endothelial cells following infection with SARS-CoV-2 [64]. Moreover, ongoing research suggests that the M1 phenotype contributes to a number of neurodegenerative diseases years following infection [65, 66]. Pro-inflammatory microglia also induce expression of genes contributing to the onset of neuroinflammation [67], further worsening inflammatory CNS outcomes.
COVID-19 AND AD
AD is the most common form of dementia [68]. The main pathological hallmarks of AD include the accumulation of extracellular amyloid plaques composed of amyloid-β (Aβ) proteins and accumulation of intraneuronal neurofibrillary tangles (NFTs) of aggregated hyperphosphorylated tau [69]. One of early pathological markers of AD, oligomeric Aβ accumulation in the brain, occurs more than 20 years prior to the initiation of clinical symptoms [70]. However, other markers of dementia are also present by the time of onset of cognitive impairments, including the accumulation of tau, glucose hypometabolism, and synapse loss [71]. Nonetheless, the aging demographic of patients likely to develop AD but prior to the onset of symptoms, makes them particularly vulnerable to severe COVID-19 outcomes, potentially exacerbating issues of comorbid dementia [72]. Interestingly, emerging reports on patients diagnosed with AD suggest that they are at an increased risk of severe complications and mortality from COVID-19 [73]. Perhaps in diagnosed individuals, the cumulative effects of existing pathological neuroinflammatory hallmarks and COVID-19-mediated responses lead to worsened outcomes post-infection.
Recent research has shown that neuroinflammation is thought to play a critical role in AD pathogenesis. In the context of COVID-19 infection, aberrant immune responses and resulting inflammation are said to advance degenerative outcomes and increase susceptibility to AD [74] (Fig. 2). Significant commonalities between COVID-19 and AD include ACE2 receptors and various markers of inflammation including IL-1, IL-6, and the APOE4 allele [75]. Furthermore, in the context of COVID-19 mediated neuroinflammatory responses, resulting biomarkers also observed in AD can be further classified according to the ATX(N) framework: “A” represents biomarkers of Aβ, “T” for values of tau, “X” for newly added biomarkers of inflammatory responses and neuroinflammation, glial cell responses, synaptic dysfunction, vascular pathologies, and other pathological changes observed in AD, and “N” for other neurodegenerative biomarkers [76].
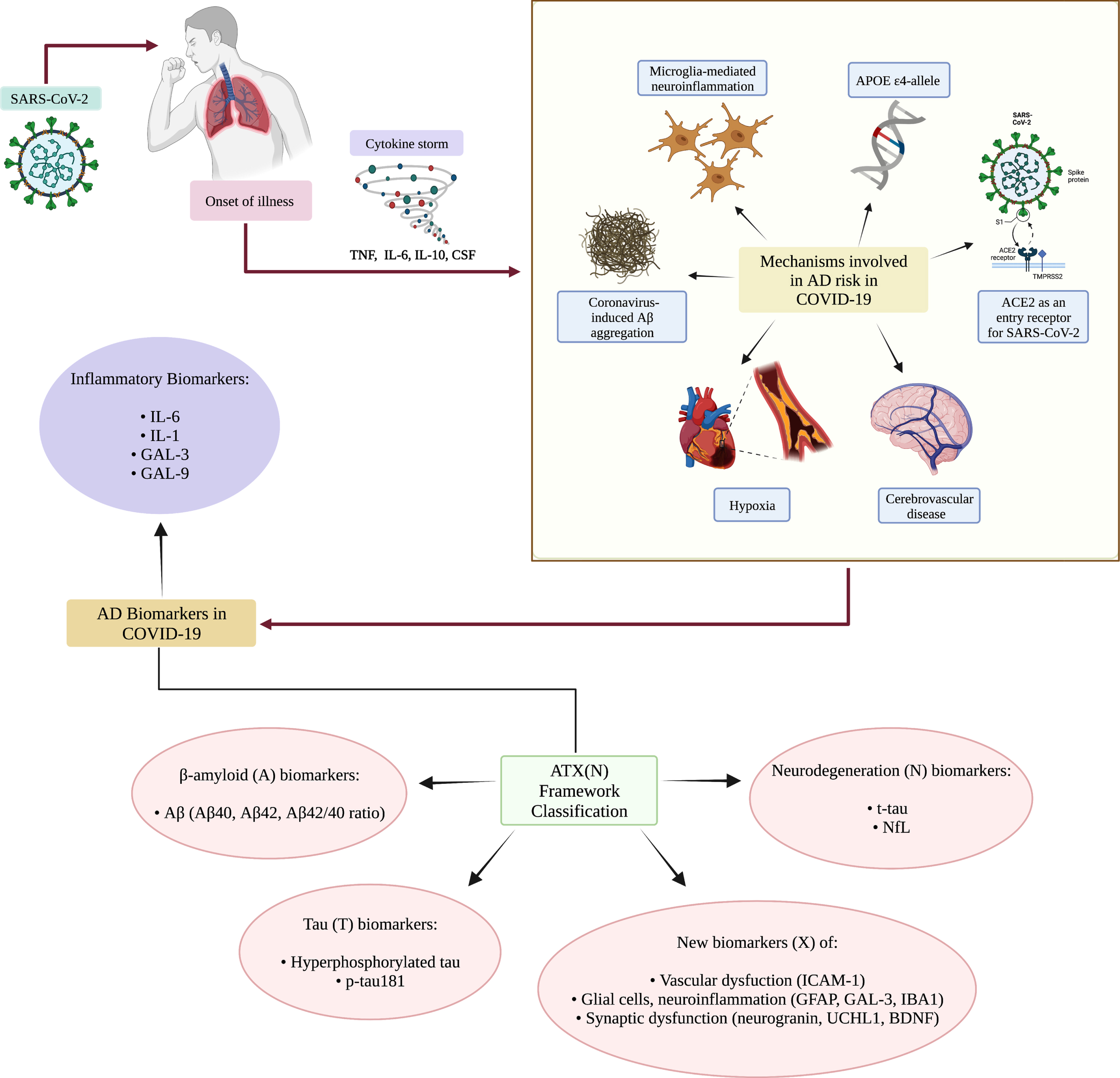
Mechanisms by which COVID-19 may be a risk factor for AD. Upon infection with SARS-CoV-2, a cytokine storm may be initiated, leading to the production of pro-inflammatory cytokines. Through various means, COVID-19 could lead to neurological manifestations and increased risk of AD. Some of the mechanisms by which COVID-19 may contribute to AD include the APOE ɛ4 allele which is a risk factor for both severe COVID-19 and AD. The genotype is linked to an increased susceptibility to Aβ fibril accumulation and oligomer formation, production of NFTs, neuronal death and oxidative stress. Microglial-mediated neuroinflammation because of the pro-inflammatory systemic response to SARS-CoV-2 can contribute to neural damage. Elevated levels of ACE2 as an entry receptor for SARS-CoV-2 adds to an elevated viral load and increases levels of Aβ and tau. Hypoxia or vascular deficits resulting from infection may also be a mechanism underlying cognitive deficits, and potentially lead to tau hyperphosphorylation and formation of NFTs. Coronavirus-induced Aβ aggregation may also be a mechanism by which COVID-19 augments the risk of AD, resulting in Aβ neuronal accumulation and synaptic loss. Lastly, cerebrovascular disease resulting from COVID-19 may explain coagulation abnormalities, damaged cerebral blood vessels, vascular dysfunction, and brain ischemia. These aforementioned mechanisms then cumulatively lead to the clinical observation of AD biomarkers in COVID-19 patients, including those of inflammation and other markers categorized within the ATX(N) framework.
MECHANISMS INVOLVED IN AD RISK IN COVID-19 PATIENTS
Coronavirus-induced Aβ aggregation
One of the most widely studied hypotheses for AD is the Aβ hypothesis; accumulation of Aβ generated via β- or γ-secretase-mediated cleavage of amyloid-β protein precursor (AβPP) in the brain results in the formation of neurotoxic pathological oligomers and senile plaques [77]. During severe COVID-19, a cytokine storm causes systemic inflammation resulting in disruption in the BBB and induction of the M1 phenotype of microglial cells. Then, arising from the massive release of pro-inflammatory cytokines such as IL- IL-1β, IL-6, IL-12, and TNF-α [78], microglial cells lose their capacity to effectively phagocytose Aβ, thereby enhancing Aβ oligomerization and aggregation [79]. Moreover, systemic release of pro-inflammatory cytokines mediates synaptic defects, which may additionally play a role in accelerating the neurodegeneration of AD [78]. Thus, the inflammatory response to the virus in addition to an increase in BBB permeability also exacerbates neuroinflammation and accelerates neurodegenerative outcomes [80]. In a recent study by Chiricosta et al., SARS-CoV-2 was found to increase levels of Aβ-mediated neurotoxicity and aggravate AD pathology [81].
There may also be a role for Aβ as an antimicrobial peptide in response to SARS-CoV-2 entry into brain. The combined cumulative effects of dysfunctional microglial-mediated clearance of Aβ and the role of Aβ as part of the brain’s immune response results in activation of the Aβ cascade with subsequent amyloid misfolding [82]. However, further research to define the precise role of Aβ in response to SARS-CoV-2 is critical in both understanding the mechanisms involved in neurodegeneration following COVID-19 and deriving targeted therapeutics.
Prior to the initiation of the cytokine storm, IFNs mediate the initial response to viral infection [83]. Interestingly, IFNs have demonstrated involvement in AD, particularly through their activation of microglial cells in the pro-inflammatory response [84]. Research by Roy et al. found that type 1 IFN responses are upregulated in AD and correlated with the extent of disease pathology [85]. Independent from the animal model of AD used, they found that type 1 IFN pathway activation resulted in accumulation of Aβ plaques, suggesting that type 1 IFNs are critical mediators of neuroinflammation in AD models [85]. Furthermore, they also found plaque-associated microglia to be a driver of neuroinflammation [85], demonstrating the role of IFNs including IFN-β in synaptic degeneration and resulting cognitive impairment [86]. It can therefore be hypothesized that in elderly populations afflicted with COVID-19 infection, their impaired IFN production contributes to a loss of immunoregulatory function. As a result, there is aberrant IFN signaling and consequential activation of the microglial M1 degenerative phenotype that induces downstream pro-inflammatory responses in addition to IFN-mediated activation of the complement cascade [85]. Collectively, these diverse immune events contribute to downstream synaptic loss and neuronal accumulation of Aβ, leading to the neuropathogenic processes associated with AD.
Microglia-mediated neuroinflammation
Neuroinflammation is recognized as a core feature of AD. In post-mortem brain analyses of fatal COVID-19 patients, severe microglial brain activation was reported [87]. In addition, SARS-CoV-2-mediated cytokine storm [88] compromises BBB endothelial function, resulting in the emergence of disease-associated microglia [89, 90]. Elevated levels of pro-inflammatory cytokines released from M1 activated microglia include IL-1β, IL-6, and TNF-α [91], which exacerbate neuroinflammation and have the capacity to contribute to neurodegenerative progression in AD [92]. When comparing serum between AD patients to healthy controls, elevated levels of IL-6 and TNF-α were detected [91]. Moreover, cognitive capacity of AD patients was reported to be negatively correlated with plasma levels of IL-6, suggesting that higher levels of IL-6 in AD may contribute to abnormalities in cellular processes mediating neuropathology [93]. Further highlighting the role of inflammatory cytokines, Bialuk et al. demonstrated that IL-6 knock-outs exhibited enhanced cognitive performances in numerous behavioral assessments [94].
The cytokine storm of severe COVID-19 results in the release of pro-inflammatory cytokines (see Fig. 2); increased levels of IL-1, IL-6, IL-8, IL-10, and TNF-α have been detected in patients’ cerebrospinal fluid (CSF) [95, 96]. Elevated levels of IL-1 and IL-6 in COVID-19 patients correlate with a worse prognosis, particularly from a CNS perspective [96, 97]. Similar to AD, age is likewise a risk factor in SARS-CoV-2 lethality [98]. In the aged lung, there is enhanced activation of the nucleotide-binding domain and leucine rich repeat containing family, pyrin domain containing 3 (NLRP3) inflammasome. As a result of NLRP3 aberrant activation, downstream levels of caspase-1, IL-1β, and IL-18 increase [98]. Similar inflammasome activation has been implicated in AD progression and pathogenesis through dysfunctional microglial clearance of Aβ [66, 100]. It is therefore reasonable to hypothesize that the inflammatory response triggered by SARS-CoV-2 in brain may lead to enhanced neuroinflammatory processes and progression of neurodegeneration, and ultimately to an increased risk for AD [101].
CANDIDATE MECHANISMS OF AD RISK IN COVID-19 INFECTION
ACE2
ACE2 is a peptidase involved in regulating the renin-angiotensinogen system (RAS) [102]. ACE2 is highly expressed in the brain in areas associated with AD pathogenesis including the temporal lobe and hippocampus, specifically on neurons and microglial cells [103]. SARS-CoV-2 uses ACE2 as an entry receptor on target cells through interactions of the receptor binding domain of the viral subunit 1 (S1) spike protein. Transmembrane protease serine type 2 (TMPRSS2), then cleaves the S protein into its S1 and S2 subunits, allowing for the fusion of viral envelope and cell membrane and subsequent release of viral ssRNA genomic material into the cytoplasm [104, 105]. The route of SARS-CoV-2 entry and COVID-19 organ-specific pathogenesis is therefore dependent on the expression levels of ACE2.
Induction of ACE pathways increases levels of angiotensin II in the brain, also contributing to oxidative stress, neuroinflammation, and reduced cognition. Although postmortem analyses of AD patient brains revealed an elevated expression of ACE2 receptors [106] and noted higher levels of the ACE2 gene to be correlated with severity of AD [107], levels of ACE2 activity are indeed reduced in AD in association with increasing levels of Aβ and tau [108]. Similarly, an in vivo study found higher levels of the ACE2 gene in brain samples in an AD mouse model [107]. To ameliorate the cognitive symptoms of AD, ACE2 therapeutics have been suggested despite their consequence of increasing levels of ACE2 [109]. Interestingly, increased ACE2 expression has also been reported to be a contributing factor to an elevated viral load in AD patients [106] and may exacerbate consequent neurological outcomes following COVID-19 infection. While neurodegenerative diseases are thought to be linked to an enhanced susceptibility to COVID-19, perhaps it can be hypothesized that in non-AD patients with higher viral loads, SARS-CoV-2 binding to ACE2 and subsequent ACE2 downregulation may precede Alzheimer’s-like pathology.
Moreover, polymorphisms in the human genome, specifically in that of the ACE2 gene may explain the differences in COVID-19 symptomatic and morbidity outcomes [110]. For instance, the ACE deletion-allele frequently observed in elderly AD patients [111], is correlated with decreased expression of ACE2 and therefore found to be protective against COVID-19 [112]. Nonetheless, numerous other variable polymorphisms of RAS components resulting in altered ACE2 gene expression and subsequent COVID-19 presentation have also been associated with AD and similar neurodegenerations [113].
APOE4
APOE is a 299 amino acid glycoprotein generated as the product of an 18 amino acid signal peptide cleavage [114–116]. APOE is generated in cell types including astrocytes and microglia [117]. Like gene variations in ACE2, the APOE ɛ4 allele is reported as a risk factor for AD, while the ɛ2 allele is protective against AD pathology [118]. The APOE4 allele is linked to an increased risk of AD and results in Aβ fibril accumulation and oligomer formation, NFT production, neuronal death, and oxidative stress [119]. It has been noted that homozygous APOE ɛ4 genotype increases the risk of AD by 15-fold [120]. This may be a result of its associated effect in AD patients culminating in reduced cerebral blood flow and neuroinflammation. In a study by Kuo et al., APOE ɛ4/ɛ4 allele resulted in an elevated risk of infection with COVID-19 [121]. Both ACE2 and APOE gene expression are elevated in type II alveolar cells of the lungs [122]. It can therefore be hypothesized that individuals with the ɛ4/ɛ4 allele are not only more susceptible to COVID-19 but also prone to the resulting long-term neurological effects and downstream AD development. Arguably, in genetically susceptible individuals who experience severe infection, COVID-19 acts as a driving force for neurodegeneration.
Hypoxia
Severe COVID-19 infection often results in ARDS [123]. This can ultimately manifest as respiratory failure or myocardial hypoxia [124], requiring hospitalization [125]. In surviving patients, post infection cognitive impairment may be explained by hypoxia or resulting vascular damage [126]. Through mechanisms which are not entirely understood [127], COVID-19-mediated hypoxia results in dysfunction of mitochondrial oxidative phosphorylation and respiration. This disabled oxygen supply then affects vascular smooth muscles and neuro-epithelial bodies [128]. Previous research by Alexander et al. observed that patients with severe brain hypoxia (including global ischemia from cardiac arrest) also exhibit impaired symptoms in many aspects of cognition as well as deficits in functional outcomes of memory and psychomotor slowing [129]. The resulting deficits in cognition may be explained in part due to the general sensitivity of hippocampal and basal ganglia brain regions in response to ischemia [130]. In particular, oxygen-sensitivity of the hippocampus may exacerbate effects on memory, thereby contributing to clinical presentations of dementia. Interestingly, hypoxia due to transient cerebral ischemia has been shown to cause tau hyperphosphorylation and NFT formation, both of which are hallmarks of AD pathogenesis [131]. Furthermore, limited postmortem data from COVID-19 patients have demonstrated the occurrence of hypoxic alterations in the brain [132], as well as vascular and demyelinating pathologies [133, 134]. In addition, a case study report found that following COVID-19, a patient developed prodromal AD as defined by a CSF biomarker panel analysis and the onset of cognitive symptoms [126]. Such findings further suggest that perhaps COVID-19 hyper-immune responses leading to ischemia may accelerate the neurodegenerative progression of AD.
Cerebrovascular disease
In addition to respiratory complications in COVID-19, acute cerebrovascular disease has also been reported [135]. In a retrospective study, Yi et al. found that COVID-19 patients demonstrated both a heightened inflammatory response and hypercoagulable state as shown by an elevated C-reactive protein level [135]. Those with underlying comorbidities such as congestive heart failure, hypertension, and diabetes are more susceptible to higher mortality outcomes and infection by SARS-CoV-2 [136]. Moreover, the study by Yi and colleagues noted that following COVID-19 infection, elderly individuals with risk factors had a higher rate of developing cerebrovascular disease (CVD) [135]. Furthermore, autopsy analyses of patients who died from COVID-19 demonstrated hyperemic and degenerate neurons [12], as well as thrombotic microangiopathy. The cumulative effect of early cerebrovascular events and coagulation abnormalities contribute to the widespread formation of thrombi in both small and large vessels [3].
CVD and AD share common risk factors and neuropathological features [137]. Shared risk factors include the APOE ɛ4 allele [138, 139], diabetes mellitus [140], hypertension [141], and age [142]. Numerous studies also implicate ischemia structural vascular pathologies in AD patients [137]. Cerebrovascular neuropathologies in postmortem AD brain analyses have identified Aβ amyloid angiopathy and arteriosclerotic small vessel disease [143], as well as frank cerebral infarcts [143–146]. Therefore, CVD is a risk factor associated with concomitant AD neuropathology arising from damaged cerebral blood vessels, vascular dysfunction, and brain ischemia [147]. Thus, COVID-19-mediated neuroinflammation in addition to the chronic side effects of pulmonary-triggered hypoxia also causes CVD [148] that in turn contributes to the amplification of AD-associated neurological processes.
BIOMARKERS OF AD/COGNITIVE DECLINE IN COVID-19 PATIENTS
Inflammatory biomarkers
Specific markers of inflammation such as IL-6, IL-1, galectin-3 (GAL3), and galectin-9 (GAL-9) have been identified as biomarkers associated with both COVID-19 and AD (see Fig. 2) [75, 149].
IL-6
As a prognostic predictor of SARS-CoV-2 progression [150], increased levels of IL-6 are correlated with increased levels of viral load and disease severity in critically ill patients with COVID-19 [96, 151]. Upon SARS-CoV-2 entry into the host, rapid viral replication induces heightened production of IL-6, thereby worsening respiratory complications. Elevated serum levels of IL-6 are a reliable predictor of respiratory dysfunction in COVID-19 [75, 152]. Interestingly in AD, elevated levels of IL-6 are also indicative of worsened disease progression and cognitive performance [93]. In IL-6 knockout mice, enhanced memory formation and retrieval, as well as cognitive flexibility have been demonstrated [94]. Furthermore, it has been demonstrated by Strafella et al. that IL-6 and IL-6R are involved in many pathogenic outcomes such as COVID-19 severity and neurodegenerative diseases [153].
As two common biomarkers of AD and COVID-19, IL-6 and its receptor IL-6R interact with glycoprotein receptors to activate downstream signaling pathways involved in immunoinflammatory modulation [154, 155]. Polymorphisms in the IL-6 and IL-6R genes influence their expression levels, binding capacity, and function to ultimately contribute to the development and progression of infectious and neurodegenerative diseases, namely COVID-19 and AD [152, 156–158]. Direct targeting of the IL-6R may therefore reduce mortality in critically ill COVID-19 patients, while also reducing neuroinflammatory and degeneration processes in AD. Clinical trial results using the IL-6R antagonists tocilizumab and sarilumab demonstrated therapeutic efficacy and increased survival in critically ill hospitalized patients with COVID-19 [159]. In a recent meta-analysis of clinical trials using IL-6 antagonists in hospitalized COVID-19 patients, a lower all-cause mortality was shown [160]. Therefore, IL-6 is a pleiotropic biomarker of COVID-19 and AD, and efforts towards identifying antagonists targeting this pro-inflammatory cytokine may provide promising therapeutic outcomes.
IL-1
SARS-CoV-2 induces a hyperinflammatory state with the release of pro-inflammatory cytokines [161–163]. IL-1, a biomarker for COVID-19 prognosis, is elevated in patients during infection onset and progression [97, 164]. Using an IL-1 receptor antagonist, anakinra, 72% of severely ill COVID-19 patients had clinical improvement in respiratory function [164]. Similarly, levels of the IL-1 family cytokines including IL-1α, IL-1β, and their antagonist IL-1Ra are increased in AD [165]. Overexpression of IL-1 in AD has been directly associated with the development of pathological alterations such as the accumulation of Aβ plaques and NFTs in neurons [166]. Further evidence of IL-1 as a driver of AD pathogenesis comes from in vivo work by Sheng et al. who demonstrated that IL-1β injections into the right hemispheres in a rat model of AD led to the development and accumulation of neurotic Aβ plaques and NFTs [167]. In contrast, inhibition of IL-1 signaling led to enhanced cognition, reduction of tau and partial decrease in specific forms of fibrillar and oligomeric Aβ [168]. IL-1 was also shown to be involved in hippocampal-dependent memory processing [169], whilst inhibition of IL-1 led to enhanced memory formation and processing [170]. Therefore, elevation in IL-1 levels following induction of COVID-19 inflammatory responses may lead to the cognitive deficits that contribute to the symptomatology of AD [171]
Galectins
Galectins are a family of β-galactoside-binding lectins that are involved in the modulation of cell death, cell adhesion [172], immune tolerance, and inflammation [173]. The most commonly expressed galectins in the CNS include GAL-3 and GAL-9 [174]. Under normal homeostasis conditions in the CNS, galectins contribute to the processes of neuronal myelination, neuron stem cell proliferation, and vesicular transport in neurons. However, in neuronal disease states and in experimental models of neuroinflammation, galectins may play a critical role as extracellular and intracellular mediators to regulate and control inflammation and to induce repair processes in injured CNS tissues [174]. In animal models of autoimmune encephalomyelitis, GAL-9 reduces autoimmune encephalomyelitis while GAL-3 stimulates inflammation. Additionally, in brain ischemic lesions, GAL-3 stimulates proliferation of microglia, whilst in models of amyotrophic lateral sclerosis it aggravates neurodegeneration [174]. Galectins, specifically GAL-3 and GAL-9, are critical mediators of homeostasis in the CNS and during neuroinflammatory processes. As galectins are involved in microglial-mediated neuroinflammation [175, 176], and microglia are subsequently responsible for the deposition of Aβ [177], under the ATX(N) framework, galectins would fall under the “X” value as biomarkers of glial cell activity and neuroinflammation.
GAL-3
GAL-3 as a β-galactoside binding to lectin results in the infiltration of neutrophils and production of pro-inflammatory IL-6 and TNF-α cytokines in cases of severe COVID-19 [178]. GAL-3 has many functions in innate immunity, particularly during inflammatory responses and induction of lung fibrosis [179, 180]. GAL-3 has been demonstrated to bind directly to pathogens through its actions as both a pattern-recognition receptor and a danger-associated molecular pattern (DAMP) [181]. It was also expressed by many inflammatory cell types, including activated macrophages [182, 183]. The GAL-3-mediated infiltration of neutrophils further contributes to airway inflammation [182, 184]; a clinical characteristic linked to disease severity and mortality in patients diagnosed with ARDS [185]. Thus, there is strong evidence for the role of GAL-3 as a prognostic biomarker of COVID-19 disease severity [186].
During severe COVID-19, levels of GAL-3 are elevated. High levels of GAL-3 were reported to promote hyper-inflammatory responses, lung fibrosis linked to alveolar destruction, hypoxia, and progression of infection [180]. Similarly in AD, levels of GAL-3 are increased, contributing to the aggregation and accumulation of Aβ plaques [187]. Additionally, in a mouse model of AD, inhibition of GAL-3 improved microglial-mediated neuroinflammatory pathogenesis [188]. Thus, based on the role of GAL-3 in viral inflammatory responses [189] and microglial-mediated neuroinflammation [188], COVID-19 patients may be at an elevated risk for the development of AD.
GAL-9
Similar to GAL-3, GAL-9 is also involved in mediating immune responses associated with viral pulmonary infection [190]. In COVID-19 patients, levels of GAL-9 were considerably elevated compared to non-infected controls. GAL-9 is also a potential diagnostic biomarker being able to differentiate SARS-CoV-2 infected from non-infected controls with a 95% specificity rate [191]. Moreover, plasma levels of GAL-9 were also positively correlated with a number of pro-inflammatory COVID-19 biomarkers in patients, thereby supporting its role as a contributor to the cytokine release syndrome in severe COVID-19 [191]. In in vitro cultures, expression of GAL-9 was upregulated in microglia, astrocytes, and oligodendrocytes [192]. From the same study, they concluded that stimulation of GAL-9 in the CNS is involved in regulating neuroinflammatory pathologies [192]. Moreover, GAL-9 has also been reported to be upregulated in patients with AD and mild cognitive impairment and was shown to be associated with reduced scores on the Mini-Mental State Examination [193]. Similar to GAL-3, severe COVID-19 and subsequent elevated levels of GAL-9 may exacerbate neurodegeneration outcomes and contribute to the risk of AD development.
BIOMARKERS OF AD NEURODEGENERATION IN SEVERE COVID-19
In a study by Frontera et al., significant elevations in biomarkers of neurodegeneration were observed in patients hospitalized with COVID-19 [194]. Markers measured included neuronal and glial specific proteins such as plasma serum total tau (t-tau), phosphorylated tau-181 (p-tau181), ubiquitin carboxy-terminal hydrolase L1 (UCHL1), glial fibrillary acidic protein (GFAP), and neurofilament light chain (NfL) [194]. The levels of biomarkers were positively correlated with increasing age and severity of infection; the highest levels of t-tau, p-tau181, GFAP, and NfL were observed in patients with encephalopathy. They also compared more specific AD pathology biomarkers such as NfL, GFAP, and UCHL1 and found that serum levels were much higher in COVID-19 patients compared to non-COVID-19 controls with mild cognitive impairment or AD [194]. Additionally, of these biomarkers, both in severe COVID-19 and AD, levels of UCHL1 [195, 196], GFAP, tau [197], and NfL [198] are also significantly elevated following microvascular changes and BBB impairment [194, 199]. Further highlighting the residual and ongoing neuroinflammation following COVID-19 infection, in the case of patients recovering from COVID-19, plasma protein markers of neuronal dysfunction such as Aβ, NfL, neurogranin, total tau, and p-T181-tau were found to be markedly elevated compared to controls [200].
In recent years, evidence for the role of chronic inflammation in AD has emerged, with recent literature in support of the reactive gliosis nature of AD [201]. The activation and proliferation of microglia in brain contribute to and exacerbate neuronal and synaptic loss, Aβ accumulation, and tau hyperphosphorylation [202]. In the case of SARS-CoV-2 infection, microglia maintain homeostatic states during viral entry in the brain, and this may ultimately lead to its hyperactivated state which is responsible for the post-COVID-19 neurological outcomes [64]. Following viral infection, release of excessive cytokines, PAMPs, and DAMPs and their binding induces activation of microglial-mediated inflammatory responses [203]. In rodents, this results in an increase in protein expression of ionized calcium-binding adapter molecule 1 (IBA1). Further evidence in support of glial reactivity in COVID-19 comes from a recent study that reported an elevated number of glia cells in the cerebral cortex of COVID-19 patients and up-regulation of Iba1+ microglia cells and GFAP+ astrocytes [204]. Additionally, a different study observed a surge in Iba1+ immune cells in the olfactory mucosa of COVID-19 patients experiencing persistent loss of smell [54].
The triggering receptor expressed on myeloid cells 2 (TREM2) is associated with AD pathogenesis, with variants in the TREM2 gene almost tripling the risk of AD [205]. Interestingly in the context of AD, TREM2 amplifies and contributes to immune inflammatory pathways [205] and affects microglial responses to Aβ and tau [206]. In COVID-19, TREM2 levels elevate and induce T cells of the lung and blood to interact with the M protein of SARS-CoV-2, thereby activating CD4+ T cells and proinflammatory responses [207, 208]. In cases of severe COVID-19, TREM2 has been associated with fibrotic processes [209]. Hence, as TREM2 is associated with both worsened pathogenic outcomes in COVID-19, and inflammatory signaling in AD, perhaps through mechanisms which are not yet delineated, exacerbated outcomes amplify neuroinflammation and ultimately increase the risk of developing AD.
In a subset of fibrogenic macrophages, the GAL-3 gene was found to be increased in association with TREM2 [209, 210]. Thus, GAL-3 has been demonstrated to play a crucial role in the microglial inflammatory response and subsequent lung fibrosis through its interaction with TREM2 [211].
Another marker of cognitive function includes brain-derived neurotrophic factor (BDNF) which is involved in the regulation of synapses [212] and in learning and memory [213]. As a mediator between inflammation and neuroplasticity, BDNF has been shown to regulate neurotransmitter release. However, as AD progresses, levels of BDNF in the brain, blood, and CSF decrease. Impaired BDNF signaling, partly in response to Aβ-induced truncation of the BDNF receptor TrkB (tropomyosin receptor kinase B) [214], is associated with Aβ accumulation, tau phosphorylation, and induction of neuroinflammatory processes [215]. In contrast, increasing serum BDNF is linked to enhanced cognition in AD [215]. In a study by Wang et al., ACE2 knockout in mice led to a reduction in BDNF levels in the hippocampus, with resulting cognitive dysfunction as compared to wild-type [216]. Alternately, inhibiting angiotensin II type 1 led to an upregulation in BDNF and subsequent prevention of memory deficits [217]. SARS-CoV-2 infection in brain results in elevated levels of angiotensin II via inhibition by ACE2 [218]. Subsequent downstream pro-inflammatory pathways then reduce levels of BDNF, yielding negative neurological outcomes including deficits in cognition [219]. Interestingly, studies have shown that levels of BDNF during COVID-19 infection correlate with disease severity [220]. COVID-19 patients with lower circulating levels of BDNF had worse outcomes [221], while recovery from infection was found to be associated with restoration in BDNF levels [220]. Thus, BDNF also serves as a common biomarker of neurodegeneration in AD and reflects the prognosis of SARS-CoV-2 infection.
In severe SARS-CoV-2 infections, elevated levels of adhesion molecules such as intracellular adhesion molecule-1 (ICAM-1) reflect inflammation and dysfunction of the endothelium [222]. Endothelial dysfunction contributes to a coagulative state, worsening COVID-19 outcomes [223]. Moreover, a separate study also evaluated the same serum biomarkers of neuronal injury and inflammation, namely NfL, t-tau, UCHL1, GFAP, and pTau-181, and found that levels significantly correlated with neurological symptoms in COVID-19 patients as compared to non-neurological symptomatic patients [224]. Thus, these findings indicate that in patients with severe COVID-19 and neurological symptoms, there is evidence for the progression of AD pathogenesis [224]. Several brain imaging studies from computed tomography (CT), positron emission tomography (PET), and magnetic resonance imaging (MRI) reported abnormalities such as white matter hyperintensities, hypoperfusion, and ischemia particularly in the cerebral hemispheres [225]. MRI scans from severe COVID-19 patients show an increase in leptomeningeal spaces; MRI of asymptomatic patients showed that some had signs of small acute lacunar ischemic strokes [226]. Furthermore, in a PET study assessing neurological and cognitive symptoms in acute COVID-19 patients, cerebral glucose hypometabolism accompanied by cognitive decline were documented [31]. A recent longitudinal study using MRI brain imaging data from UK Biobank participants, comparing COVID-19 infected and non-infected, found reductions in grey matter thickness in orbitofrontal cortex and parahippocampal gyrus regions, tissue damage in the olfactory cortex, and reduction in brain size following infection [11]. These COVID-19 associated imaging abnormalities are similar to those documented in AD.
A trial in the Netherlands has attempted to measure in vivo neuroinflammation using TSPO-binding PET employing a radioligand that targets the Translocator Protein elevated in activated microglia in recovering COVID-19 patients experiencing fatigue and cognitive deficits [227]. A similar study is using a longitudinal design with a series of serially performed assessments to generate a dataset derived from severe COVID-19 patients with delirium to evaluate neuroinflammatory and neurodegenerative complications. Assessments include repeated SARS-CoV-2 testing, systemic and central immune responses evaluated using biomarkers of CNS damage from peripheral blood and CSF, in vivo brain PET-TPSO scans, functional and structural MRI scans, and multi-domain neurocognition evaluations [228]. Ongoing longitudinal clinical trials using structural and functional imaging techniques are urgently needed to evaluate the pathological CNS changes induced by SARS-CoV-2, and to facilitate the development of targeted therapeutics.
THERAPEUTICS
Despite growing concerns and emerging cases of COVID-19 mediated neurological outcomes, there are currently no ongoing clinical trials addressing this important brain-focused phenomenon. The best therapy for such neurological sequelae is therefore prevention or early treatment, through either mass vaccination efforts or antiviral approaches using paxlovid [229], molnupiravir [230], or fluvoxamine [231]. However, vaccination is mostly effective at reducing severity and mortality outcomes while providing short-term protection against infection and may not be fully effective against newer strains of SARS-CoV-2 [232, 233]. In addition, oral antiviral therapies like paxlovid include the risk of COVID-19 rebound syndrome [234] and elevate susceptibility for resistance-causing mutations in the case of molnupiravir [235, 236].
An alternative therapeutic approach could address COVID-19-mediated neurodegeneration via targeting of neuroinflammatory mechanisms [237–240]. Building on data suggesting that BBB dysfunction is associated with neuroinflammation and AD [241], an ongoing clinical trial is using transcriptomic analyses and microglial modeling to evaluate neuroinflammation in AD to identify causative factors [242]. Moreover, an interventional early Phase 1 trial is assessing brain inflammation and cerebral amyloidosis in AD patients using PET/CT imaging techniques [243]. Although there are few trials evaluating neuroinflammation in the context of COVID-19, one ongoing Phase II/III study is attempting to quantify neuroinflammation using PET imaging in recovered COVID-19 patients in relationship to psychiatric and cognitive symptoms [227]. As the need for an immediately available therapeutic is urgent, repurposing already approved drugs may be a promising approach. Furosemide, a loop diuretic approved by The Food and Drug Administration (FDA) for the treatment of edema following congestive heart failure, liver failures, or renal failure [244], is also capable of broadly inhibiting pro-inflammatory cytokines like IL-6, IL-8, and TNF-α [245], all of which induce hypercytokinemia and pulmonary tissue destruction [246]. Based upon this, a randomized double-blinded and placebo-controlled Phase II/III trial is investigating nebulized furosemide for treatment of pulmonary inflammation and respiratory failure in hospitalized COVID-19 patients [246–248]. Interestingly, furosemide has been shown to prevent the release of proinflammatory cytokines TNF-α, IL-6, and nitric oxide, and has capacity to reduce microglia M1 phenotype while upregulating M2 [249], thereby suggesting potential as a neuroinflammatory therapeutic in AD. These data along with a number of studies demonstrating loop diuretics are associated with a reduced risk of AD and dementia [250–252], presents an interesting opportunity to develop these class of molecules to treat neuroinflammation.
CONCLUSION
Over 55 million people worldwide are living with AD [253], and 10 new million cases of dementia are reported yearly [254]. However, these statistics do not consider the potential added burden from COVID-19 neurological manifestations. Out of more than 500 million confirmed COVID-19 cases, one third of patients develop neurological symptoms in the acute stage alone, ranging from headaches [255] to neuronal degeneration, including 25% of symptoms, which are a result of direct CNS involvement [11]. Moreover, growing reports of PASC and evidence of systemic inflammation as a driver of cognitive decline and neurodegeneration [256, 257] suggest that survivors of COVID-19 are at an elevated risk of developing AD. COVID-19 is therefore an immediate threat to not only our healthcare systems, but also to the growing pandemic of AD.
Estimates from the United Nations suggest that global rates of AD may exceed 150 million by 2050 [258]. Combined with the lack of breakthrough pharmacological therapeutics for AD, and the unknown mechanisms by which COVID-19 may contribute to its development, 150 million may be a gross underestimate. Greater research is urgently needed to assess and evaluate the symptoms and neurological sequelae of COVID-19 survivors. Out of the third of survivors developing CNS complications and the recent emergence of reports from long COVID-19 patients experiencing neurological manifestations [259], how likely is it that all, or a large fraction thereof, may go on to develop AD? Is it the severity of illness or rather acute vs long COVID-19 that are the major contributors to AD? To date, no study has provided conclusive answers.
A large body of evidence has suggested an association between severe COVID-19 infection and dementia [260, 261]. Dementia patients diagnosed with COVID-19 were reported to have worsened clinical outcomes, elevated hospitalization rates, and overall higher mortality [194, 263]. While the fatality of COVID-19 is higher in dementia patients due to old age, immune dysfunction, and comorbidities, limited data exist on the risk of AD in COVID-19 patients. The continuous rising incidence and prevalence of COVID-19, combined with a dramatic rise in AD cases and mortality [264] poses significant personal and socioeconomic impacts. In 2020, the healthcare costs for AD treatment were estimated at $305 million and expected to reach over $1 trillion [265]. These values do not consider the socioeconomic and personal burdens associated with indirect costs of care such as quality of life and private caregiving [265]. Furthermore, the COVID-19 pandemic has had tremendous global economic burdens. COVID-19 as a risk factor for AD is therefore an association with great public health consequences, and which remains a critical unmet global medical need.
In addition to the lack of research on the association of COVID-19 infection and AD, limited data exists regarding the pathological mechanisms by which SARS-CoV-2 affects the CNS. However, based on current literature, mechanisms involved in the pathogenesis of AD are thought to be implicated in COVID-19, with interchangeable inflammatory and AD biomarkers being noted in recovering patients. Greater initiatives are required to accelerate research on neurological outcomes following COVID-19 and to develop preventative or therapeutic approaches to ameliorate the neurological sequelae that lead to an elevated risk for AD.