
Abstract
Advances in biomarkers, genetics, and other data used as dementia risk evidence (DRE) are increasingly informing clinical diagnosis and management. The purpose of this Mini-Forum is to provide a solutions-based discussion of the ethical and legal gaps and practical questions about how to use and communicate these data. Investigators often use DRE in research. When participants ask for their personal results, investigators have concerns. Will data that was intended to study groups be valid for individuals? Will sharing data cause distress? Debates around sharing DRE became heated when blood-based amyloid tests and amyloid reducing drugs appeared poised to enable clinicians easily to identify people with elevated brain amyloid and reduce it with a drug. Such an approach would transform the traditional role of DRE from investigational to foundational; however, then the high costs, uncertain clinical benefits and risks of the therapy led to an urgent need for education to support clinical decision making. Further complicating DRE use are direct to consumer genetic testing and increasingly available biomarker testing. Withholding DRE becomes less feasible and public education around responsible use and understanding become vital. A critical answer to these legal and ethical issues is supporting education that clearly delineates known risks, benefits, and gaps in knowledge, and communication to promote understanding among researchers, clinicians, patients, and all stakeholders. This paper provides an overview and identifies general concepts and resource documents that support more informed discussions for individuals and interdisciplinary groups.
Keywords
DEMENTIA RISK EVIDENCE (DRE) YESTERDAY AND TODAY
Dementia researchers are developing technologies that estimate risk for Alzheimer’s disease (AD) and other forms of dementia with increasing ease and precision. For example, the process of detecting brain changes contributing to AD is being transformed. Researchers used to restrict definitive AD diagnosis until after death when the abnormal forms of the proteins amyloid and tau could be identified in the brains of patients. Biomarkers make possible detection of AD during life. A biomarker (short for biological marker) is an objective measure that detects what is happening in a cell or an organism at a given moment (for further explanation, see [1, 2]). Biomarkers for amyloid and tau, derived from cerebrospinal fluid (CSF) obtained by lumbar puncture or spinal tap, or measured by positron emission tomography (PET) brain scans in living people, revealed that this brain pathology builds up decades before Alzheimer’s symptoms develop [3]. Many patients are reluctant to undergo spinal taps, and PET scans are not easily available outside of specialized dementia research and clinical settings, hence access to these tests was limited. But now there are blood test-based biomarkers for amyloid which could easily be used outside of specialty dementia clinics, and other blood-based biomarkers are emerging. Just as people can obtain information about a genetic risk for AD from companies that sell their services direct to consumers, it is technically feasible that a blood-based amyloid biomarker could similarly be made available. Dementia experts know that an Alzheimer’s diagnosis is complex and should never be based exclusively on a biomarker alone [4]. Experts also advise caution in using blood-based biomarkers while they are new, since sometimes these blood-based amyloid biomarkers yield subtly different results from more well studied biomarkers [5]. There are now drugs that can reduce brain amyloid levels (e.g., aducanumb, lecanumab, gantenerumab). Although these drugs have risks and the clinical meaningfulness of their effects may be small, the potential for slowing progression or preventing AD could increase pressure on clinicians to perform blood tests and prescribe these medications.
The story of amyloid biomarkers and therapies is just one example of why the field needs to examine how we use and share DRE to minimize risk, maximize benefits, and respect the individual needs of people living with and at risk for dementia. This process is evolving. Different forms of DRE are beginning to provide different types of potentially clinically actionable information. Some of the data researchers hold thus may be clinically meaningful to their participants. Research participants are asking for their results [6–8] and researchers are increasingly sharing these data [9, 10].
The use of DRE is also becoming more complex and nuanced. Traditionally genetics was used to identify vulnerability to dementia. Biomarkers provided additional information around the current disease state. Now, pharmacogenomics is using genetics to predict drug response (e.g., [11]). The US Food and Drug Administration (FDA) identifies multiple, diverse roles for biomarkers [12]. Because the controversial FDA accelerated approval of aducanumab for the treatment of AD is based heavily on biomarker evidence, educating patients about the risks, benefits, and limitations of the research becomes critical for informed decisions [13]. Aside from drug therapies, the predictive power of DRE also has the potential to guide clinical practice (e.g., [14]) and social policy [15] as large datasets from populations (e.g., [16–18]) offer new opportunities to learn about real world risks and protective factors.
The debates around these issues have been ongoing for many years; however, recent scientific advances raise the urgency of developing ongoing, effective, balanced, dementia risk communication tools and guidance. For example, in this Mini-Forum we discuss how a blood test predicting brain amyloid now makes this biomarker easily available but requires judicious use [19], and more of these tests are under development. This introduction to the Mini-Forum has a dual purpose: 1) to define commonly used concepts, acronyms, and to identify key foundational references and 2) to provide a context for the papers in the Mini-Forum on Communicating and Using Dementia Risk Evidence. The most effective guidance will likely come from multidisciplinary perspectives and open, and fluid, consultation among researchers, clinicians, ethical, and legal scholars, and informed consumers and stakeholders. Contributions to this Mini-Forum and many of the ideas presented here come from members and subgroups of the Advisory Group on Risk Evidence Education for Dementia (AGREEDementia.org), an open working group that discusses and identifies issues related to this topic [20], and papers submitted to Ethics Review that were relevant. There is a strong emphasis here on US guidelines. Models from outside the US come from excellent guidance [21, 22] and examples of clinical trial registries that handle DRE effectively [23, 24]. There was also a great example of a survey on the use of CSF in mild cognitive impairment in Germany [25]. But more work is needed. In general, this Mini-Forum focuses on the perspectives of the research and clinical community, but it is important to consider the perspectives of the research participants, their support partners and families, and the community receiving care. We begin this Mini-Forum with a proposal from the AGREEDementia stakeholder group [8]. Finally, in this overview we often use the term DRE rather than restricting discussion to biomarkers and genetics, the classical indicators of dementia risk. The US FDA biomarker guidance indicates biomarkers do not include measures of “how an individual feels, functions, or survives” [12]. We believe it is important to not exclude what Au and colleagues have termed “digital biomarkers” of behavior (e.g., actigraphy to derive sleep or activity, audiometric analyses of speech, or video gait analyses) which likely have parallels to classical biomarkers and their own, additional, ethical issues [26].
A RESEARCH PARTICIPANT’S BILL OF RIGHTS
This Mini-Forum begins with a strong statement from a group of stakeholders (including people living with and at risk for dementia and advocacy organizations that support them) that researchers should increase sharing of individual level data with their participants [8]. The importance of planning for communicating unexpected, incidental findings that reveal urgent care needs has long been recognized (e.g., [27–29]). This paper revisits the issue of the return of individual-level research results in the context of current innovations in Alzheimer’s research. In general, sharing information that identifies urgent treatment needs is mandatory. Sharing non-urgent information that is clinically useful for guiding therapy is always encouraged. The authors suggest that for many participants, clinically valid (i.e., having scientific evidence of validity and reliability) results of increased DRE are personally valuable and inform life decisions even if there is no established treatment. This group also advises that interested participants could be more meaningfully engaged as partners in research if they received their own, “cutting edge” data, that do not yet have evidence of clinical validity, if there are appropriate safeguards and education.
GENERAL ETHICAL PRINCIPLES APPLIED TO DRE
Ethical principles (autonomy, beneficence, maleficence, justice) provide a powerful framework for the effective use of DRE. Whereas there are more extensive reviews of ethics applied to DRE (e.g., [30]), here we provide examples and these themes occur throughout the Mini-Forum.
Clinicians and society also have a duty to support
ETHICAL TOOLS/APPROACHES FOR DRE COMMUNICATION
Ethical principles often conflict, and autonomous choices depend critically on deeply personal values related to risks and benefits, both anticipated and unanticipated. These discussions take time and expertise. Specialists such as genetic counselors and specialty dementia clinics are ideal for supporting these discussions, but there may be insufficient numbers of clinicians to fulfil the need. In this Mini-Forum, Arias et al. [51] interviewed alternative front line care providers, geriatricians, about APOE genetic testing. Their main concern was the clinical utility of these tests. Also in this Mini-Forum, we describe a structure for an inclusive, multidisciplinary, ongoing, discussion forum, AGREEDementia that is helpful in developing education and other solutions to meet the need [20].
Decision aids/tools
One way of efficiently guiding informed choice is through educational support materials for patients and stakeholders before and after tests. A decision aid occurs before DRE testing to help people decide whether they want to collect or learn DRE. These decision aids pose questions and provide information. Another aid is provided after DRE are collected and interpreted to describe the meaning of results. Both of these components support autonomy in the right to know or not know, and to understand the limitations and implications of that DRE for other decisions. Examples have been published [52–54] and are available from NIA and on the AGREEDementia website [55]. Supporting these decisions becomes complicated in the context of people living with dementia, particularly in longitudinal studies where decision making capacity is expected to decline. In this Mini-Forum, Largent et al. [56] explain why identifying a study partner to support such discussions as learning DRE is helpful as cognition declines in participants.
Control over digital privacy and the medical record
The US 21st Century Cures Act enables full access to the medical record and thus introduces both the opportunity to use DRE clinically, but also the risk for misinterpretation and misuse of DRE [31, 57]. In this Mini-Forum, Lerner [58] cautions that clinicians should consider carefully why they are collecting DRE and they also have a duty to protect patients who have biomarker or genetic data in their medical record (e.g., when participating in a research study) from the false conclusion that this information is sufficient to constitute a diagnosis. Patients may access biomarker results in their medical record before a clinician can explain these results. One solution some centers have used (e.g., Washington University) is to delay access to this information until the clinician discusses the results with the patient (for a discussion on this practice, see [57]). People sometimes choose to undergo direct to consumer genetic testing to learn results. In this Mini-Forum, Zallen has also found that they also choose to obtain the tests outside of the healthcare settings to keep them out of their medical record [59]. Digital biomarkers can also have particular privacy risks, such as with geolocation or proximity indicators (i.e., when the individual is near another person identified by their unique phone). For information-rich datasets such as MRI, there is the potential for identification with sufficiently powerful algorithms [60], hence there are often additional protections such as warnings during informed consent or anonymization software.
CRITICAL CONCEPTS AROUND DRE
The issues in this section are relevant for deciding to undergo testing and understanding test results. Part of what causes conflict around communicating and sharing DRE is that, with evolving science, there are levels of uncertainty. Measures used for clinical decision making must be accurate. Specifically, they must be reliable (e.g., if measured on two consecutive days results will be comparable) and valid (i.e., show evidence that they measure what was intended or predict an outcome). Even if a measure is accurate, there are other reasons for uncertainty in conclusions which we describe below.
Accuracy of results
It is important to discuss with stakeholders and patients the accuracy and level of uncertainty of DRE. This information is particularly important for genetic testing where direct to consumer products are available [41, 61]. Clinical Laboratory Improvement Amendments (CLIA) certified test results adhere to FDA governed clinical standards. In contrast Laboratory Developed Tests (LDT) do not adhere to these standards. The FDA discusses when LDT are appropriate and when there are risks for relying on them [62]. FDA approval may only hold for specific genetic tests but deriving additional data may not be within the approval. For example, 23 and Me has FDA approval for APOE genetic testing. Among other requirements, this approval mandates that related education and information on support be provided. However, this company also enables consumers to take their genetic data to less well validated, online services (e.g., Promethease) to explore other genetic mutations and these results do not have the same rigorous validation. Serious and unnecessary distress could result if, for example, an individual receives erroneous information that they carry a penetrant mutation for a serious illness (Jill Goldman, personal communication).
Genetic penetrance versus susceptibility
People considering genetic testing must understand that mutations vary in how consistently they lead to a disease. The classic example is Huntington’s disease, in which an expansion in the HTT gene guarantees developing the illness. The term “pathogenic variant” is preferred when referring to the penetrant mutations associated with frontotemporal dementia [63]. For highly penetrant pathogenic variants, genetic counsellors are critical to help people decide whether they wish to know, with high certainty, that they and their family members will inherit the disease [64, 65]. In contrast, for genetic variants that convey mild to moderately elevated risk and only increase susceptibility to dementia, the general guidance is that sharing this information with people without symptoms is not useful since predictive certainty is much lower [39, 46], and elevated genetic risk for dementia can be stigmatizing (but see [8] in this issue regarding personal utility). For example, the ɛ4 allele of APOE, the gene encoding apolipoprotein E, is associated with higher risk of AD dementia, but many APOE ɛ4 carriers never develop dementia. The calculus becomes different when there is clinical utility. APOE ɛ4 carriers have a higher risk of amyloid-related imaging abnormalities (ARIA) related to aducanumab and other amyloid immunotherapies (including gantenerumab, lecanamab, and donanemab), so that APOE ɛ4 carrier status may be relevant to clinical decision making regarding these treatments [42]. The US National Human Genome Research Institute (NHGRI) has educational resources and the American College of Medical Genetics and the National Society of Genetic Counselors produce guidelines regarding disclosure of genetic results which identify important concepts [39, 63]. There are standard conventions for referring to genes and the proteins they encode which we provide for a gene strongly associated with risk for AD, APOE, in Table 1 (courtesy of Suzanne Schindler).
Biomarkers: Use and interpretation
Whereas it may be tempting to assume biomarker tests will always yield simple, dichotomous (positive/negative) answers, with new biomarker tests, interpretations may need to be nuanced as their relative strengths and weaknesses are uncovered. For example, different biomarkers for AD (amyloid, tau, and neurodegeneration/hippocampal atrophy) reveal pathology at different disease stages and the A/T/N framework that leverages these multiple indicators of disease progression may ultimately serve as a method of describing these stages [66]. At the same time, different modalities for measuring these biomarkers (e.g., CSF, blood, PET, MRI) and types of measures (e.g., p-tau181, p-tau217) must be validated across different disease stages (e.g., [67]). A Roadmap and glossary of terms for validation of biomarkers has been addressing these issues [67, 68] built upon the approach of oncology [69].
Biomarker context of use and appropriate use criteria
Clinicians typically use AD biomarkers in accordance with the recommendations of expert panels that define AUC (e.g., [5, 42–44]). The FDA oversees formal approval of biomarker tests. This approval may increase the likelihood of insurance reimbursement for AD biomarker testing. Although several amyloid PET tracers are FDA approved, amyloid PET is not reimbursed outside of research studies. The FDA also approves the purpose for which they are used (although tests are sometimes used “off-label”, i.e., outside of FDA approved indications). The term describing approved indications is context of use (https://www.fda.gov/drugs/biomarker-qualification-program/context-use) (Fig. 1) [12]. For example, biomarkers might be used in guiding treatment selection, monitoring treatment response, supporting early detection, enabling early intervention and prevention, and as surrogate endpoints [12]. Biomarkers indicating the presence of amyloid in the brain can be helpful in dementia diagnosis. For example, if a patient with memory impairment has biomarker evidence of amyloid in their brain, this finding may support AD as the etiology of memory impairment. A prognostic biomarker needs to be predictive. For example, an amyloid PET scan or blood test may not be an appropriate prognostic test in a person without symptoms since not everyone with a positive brain amyloid scan develops dementia and hence in patients without clear evidence of cognitive dysfunction there is a reluctance to use this information clinically.
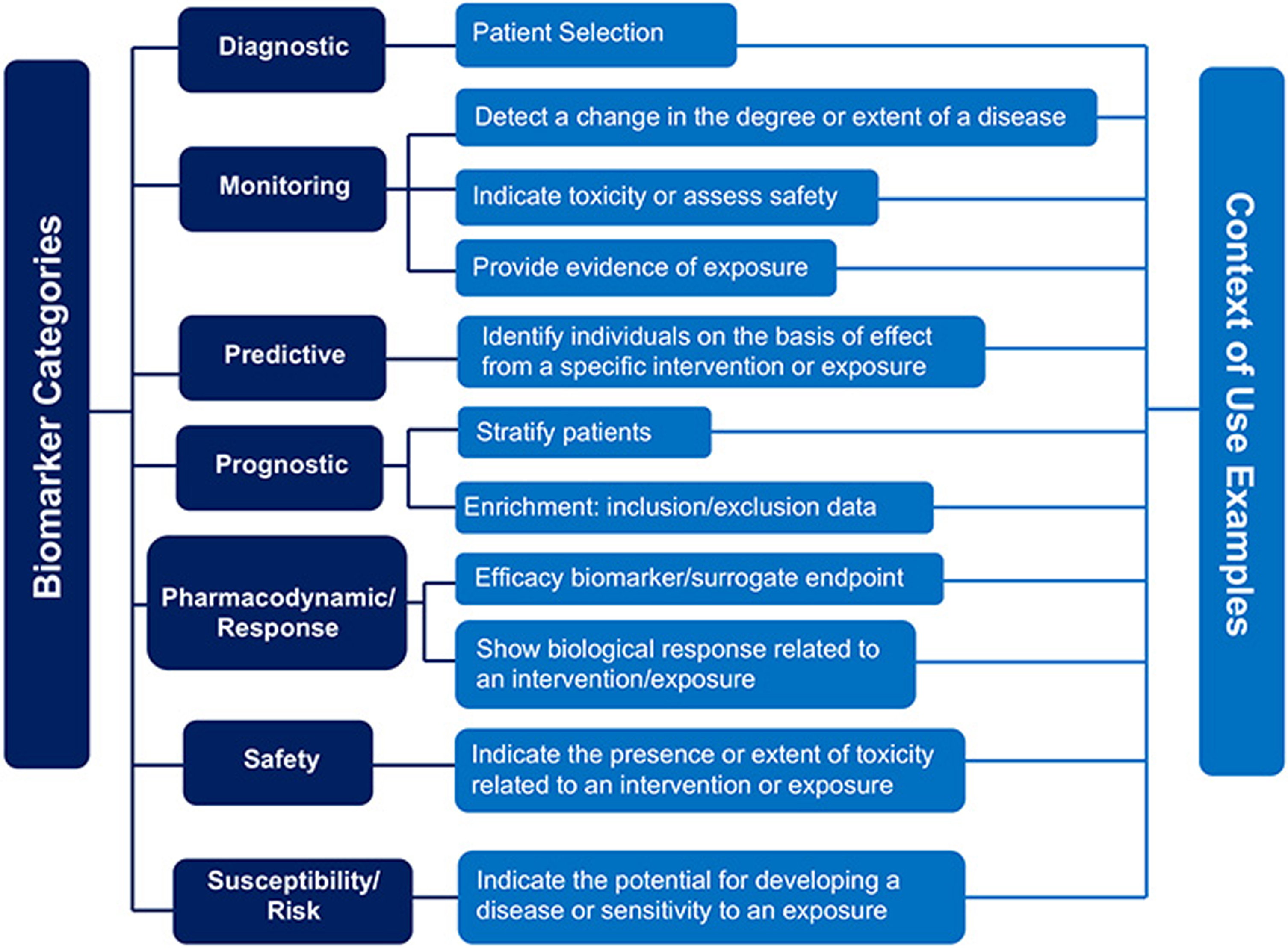
FDA Guidance on context of use (https://www.fda.gov/drugs/biomarker-qualification-program/context-use).
The surrogate endpoint use of biomarkers is critical to understand for Alzheimer’s-related medications that target amyloid or tau reduction. Whereas clinical trials ideally target clinical outcomes such as making patients feel better, slowing cognitive decline, or living longer, these outcomes may take too long to be feasible in a trial. A biomarker could serve as a surrogate endpoint, a substitute for a clinical endpoint, but should be mechanistically related to the disease and should correlate with a direct measure of clinical endpoint. For example, a reduction in brain amyloid as measured by PET is claimed as evidence that a drug has affected AD [70]. However, the evidence supporting a clinical benefit associated with amyloid reduction is inconsistent (e.g., [13, 72]). In this Mini-Forum, Schindler suggests that one potential reason for the difficulty in finding relationships between amyloid levels and cognition is that the relationship is nonlinear [73]. She also suggests that understanding this relationship could someday give patients an answer to an often-asked question of how long they have before their cognition declines. Also in this Mini-Forum, Parra suggests that leveraging subtle and refined brain-behavior relationships will support more sensitive associations between measures of brain pathology and cognition [74]. A previous Mini-Forum on digital cognitive testing further demonstrates examples of approaches to improve sensitivity to cognitive decline that could support more sensitive associations to measures of brain integrity [75].
SOME CURRENT CONTROVERSIES
Biomarkers in cognitively normal individuals
Clinical biomarkers such as measures of elevated amyloid are typically only collected clinically in people with cognitive dysfunction because this information has benefits such as supporting differential diagnosis or indicating they may be appropriate candidates for amyloid lowering treatments. For people with no symptoms, biomarkers may not provide clear information at an individual-level about risk for dementia. Because of the insidious onset of most dementias, the distinction between normal and symptomatic is not binary. Whereas neuropsychological tests often have appropriate normative data, the majority of biomarker studies are derived from highly educated, Caucasian individuals. Still more problematic is that individuals with subjective cognitive decline are at high risk for subsequent cognitive decline [76]. These individuals are not satisfied with being told the disclosure of biomarker results is not relevant for them. The AGREEDementia stakeholder working group states that people, regardless of cognitive status, should be able to receive these data and that for people without symptoms these data are personally valuable in helping them make plans [8]. In this Mini- Forum, the AGREEDementia working group that focuses on applications to people without symptoms provided a thoughtful discussion on issues to consider and language to use in applying biomarkers to people without symptoms [77].
Diversity, social determinants of health, comorbidities
Ethical concerns loom large as DRE is used to guide policy. In this Mini-Forum, Karikari [78] explains that with the advent of blood-based biomarkers, there is great potential for expanding access to minoritized populations and developing countries. However, even for established laboratories, there may be uncertainty in classifying level of risk in the context of various individual differences due in great part to the failure of dementia research as a whole to recruit cohorts that reflect the racial and ethnic and socioeconomic diversity, particularly in the US. For example, biomarker levels sometimes show differences across cohorts that differ in racial ethnic status, comorbidities, and social determinants of health, hence predicting dementia may need adjustments that are not yet well worked out [79–83]. In this Mini-Forum, Daly et al. [15] urges that since treatments that target brain pathology are not yet demonstrated to be therapeutically valuable, it is important to allocate resources toward risk reduction that maximizes the overall societal good. Intervening in populations where social determinants of health are disproportionately contributing to dementia would thus be an opportunity.
How to implement biomarker-based criteria for therapies is also problematic in light of the lack of experience with diverse cohorts. For example, if biomarker levels erroneously suggest greater brain health in a certain race (e.g., lower amyloid levels), the cut point for access to therapies may be higher, and there may thus be diminished access to the biomarker-related treatments [84]. Conversely, a lower bar may lead to over treatment. Recent studies suggest that some but not all dementia biomarkers may be resilient against these effects [85]. Regardless, there is a great need for clinical trials in diverse cohorts to ensure therapeutic results generalize [86].
OVERVIEW OF MINI-FORUM
These basic concepts provide a framework for appreciating the papers in the Mini-Forum on the communication and use of DRE in light of recent advances. The Mini-Forum begins with Walter and Taylor et al. [8] who put forward a proposed Research Participant’s Bill of Rights advocating that participants in dementia research should have increased access to their individual results. Karikari [78] describes how ease of use in blood-based biomarkers makes DRE more widely available in under-resourced settings, thus supporting international justice through biomarker supported diagnosis. Daly et al. [15] advise to be strategic about how resources are deployed to at-risk populations to provide the most societal good, maximizing beneficence.
With rapid advances there must be careful attention to the quality of evidence surrounding DRE as it accumulates and outreach supporting appropriate use. Galasko et al. [19] describe how novel blood-based biomarkers must be used carefully to ensure their results are valid in the settings they are used. Effective analyses enhance the power of DRE and Schindler shows how it is possible to use these data from biomarkers to answer meaningful questions such as “How long do I have” before developing dementia [73]. Effective cognitive measures are also crucial for validating biomarkers and Parra [74] also points to the importance leveraging cutting edge neuroscience to improve the sensitivity of cognitive measures to brain dysfunction.
Educating, engaging, and understanding the perspectives of diverse clinicians, people living with and at risk for dementia and their advocates, and the public is also vital for supporting effective use of DRE. Rosen et al. [20] describe a group (AGREEDementia) that facilitates ongoing discussion, education, and dissemination. Arias et al. [51] describe an example of how to survey the perspective of frontline clinicians, geriatricians, about DRE. Walter et al. [34] describe a method of engaging people living with dementia in attending scientific conferences. Largent [56] suggests including study partners of people with increased risk of dementia before cognitive decline progresses to ensure their wishes are protected in longitudinal studies.
Finally, understanding the context surrounding how and where DRE is represented is important for representing its significance. Lerner [58] cautions that failure to provide context for DRE, such as when biomarker data from research participants are in the medical record, can lead care staff to infer dementia diagnosis from an individual biomarkers. Zallen [59] observed that keeping DRE out of the medical record while learning results is a motivator for people pursuing direct to consumer testing. Finally, Mozersky et al. [77] caution that FDA approvals of novel amyloid reducing medications to treat mild AD could alter the disclosure of amyloid positive biomarkers in research on people without symptoms. They provide examples of language that could be used to facilitate this communication.
CONCLUSIONS
This discussion provided a basic explanation and context for the Mini-Forum on ethical, legal, and practical issues on communicating and using DRE. The increased availability of DRE such as through blood-based biomarkers and direct to consumer genetic tests could transform diagnosis and care; however, use must be judicious. DRE varies in predictive ability and utility for individuals. We discussed how a general ethical framework can be related to these decisions and, in our experience, respect for autonomy and differences in values facilitates consensus. Decision tools are efficient methods for helping improve informed choices in patients and other stakeholders, particularly when expert clinician time is limited. Presently, the most effective use of DRE is where it can contribute to diagnosis and care in people with symptoms; however, we provide information on off label use in people without symptoms [77]. Once DRE have been collected, it is vital that they not be used as short-cut for a comprehensive and careful evaluation of contributors to symptoms, particularly treatable conditions. With advances in the predictive ability of DRE and therapies we hope someday, DRE can enable presymptomatic detection and prevention of dementia. In the face of rapidly evolving innovation, making decisions around appropriate context of use, assuring that effective communication leads to understanding, and informed decision making will be challenging. Above all, there is a need for avoiding paralysis of indecision in the face of uncertainty since the degenerative process of dementia waits for no one, especially not the people living with dementia.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0722r1).