
Abstract
Alzheimer’s disease (AD) is one of the most common neurodegenerative diseases worldwide. The accumulation of amyloid-β (Aβ) protein and plaque formation in the brain are two major causes of AD. Interestingly, growing evidence demonstrates that the gut flora can alleviate AD by affecting amyloid production and metabolism. However, the underlying mechanism remains largely unknown. This review will discuss the possible association between the gut flora and Aβ in an attempt to provide novel therapeutic directions for AD treatment based on the regulatory effect of Aβ on the gut flora.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder mainly characterized by progressive loss of cognitive ability [1]. The accumulation of amyloid-β (Aβ) into plaques in the brain tissue and neurogenic fiber tangles due to tau protein misfolding are important pathological features of AD [2]. At present, AD remains one of the major global health problems affecting human health. AD affects about 6.2 million Americans ≥65 years of age in the United States, and the number of AD patients is estimated to reach 13.8 million by 2060 if no effective intervention can be offered in time. According to the official statistics, 214,499 individuals died of AD in 2019, ranking the sixth leading cause of death in the United States. From 2000 to 2019, the death rate from stroke, heart disease, and HIV was reduced, whereas the death rate from AD rose by more than 145% [3]. Therefore, there is an urgent need to find new effective therapeutic strategies.
The enteric nervous system (ENS), known as the second brain, is closely associated with the typical pathological features of AD. It was found that the deposition of Aβ, phosphorylation of tau protein, and the increased level of total tau protein were observed in the myenteric plexus of APP/PS1 [4]. Meanwhile, it was accidentally found in autopsy that Aβ was present in the gut of AD patients but not in the gut of non-AD controls [5]. Meanwhile, Puig et al. [6] suggested that Aβ may first accumulate in the enteric nervous system, leading to immune cell activation and neuronal dysfunction in the digestive tract, and later they analyzed the ileum of amyloid precursor protein (AβPP)/progerin 1 (PS1) mice and C57BL/6 (WT) mice, finding that the levels of IgA, Aβ, and protein cleavage enzyme (BACE) in the lumen of AβPP/PS1 mice were increased, and the corresponding intestinal water levels of Aβ1 - 40 in tract lysates, and Aβ1 - 40 and Aβ1 - 42 in feces were also increased. There are also studies saying that human Aβ is expressed not only in the mouse brain but also in the intestinal tissue [7]. They then injected Aβ1 - 42 oligomers into the gut of ICR mice and found that these oligomers first migrated through the submucosa to nearby areas and then internalized into cholinergic neurons in the gut. In vivo imaging and tracking revealed that Aβ migrated to the gastric body and proximal colon, and partially distributed in the gastric fundus and jejunum within a month. A year later, beta-amyloidosis developed in the vagus nerve and brain neurons, with Aβ deposits [8]. During this period, Manocha et al. [9] proposed that the enteric nervous system produces Aβ in intestinal diseases, and they experimented on C57BL/6 (WT) male and female mice of different months of age with APP/PS1 mice and NL-G-F (App NL - G - F) mice, which are both of Alzheimer’s disease model mice. Aβ plaque deposition was first observed in the brain of AppNL - G - F mice. When at 3 months of age, the female AppNL - G - F mice exhibited decreased intestinal motility and increased intestinal permeability in female APP/PS1 mice. However, both male and female APP/PS1 and AppNL - G - F mice had elevated levels of colonic lipid carrier protein 2 mRNA and insoluble Aβ1 - 42. However, a large number of other studies showed that the occurrence of intestinal diseases was closely related to the imbalance of intestinal flora [10, 11]. Therefore, intestinal dysbiosis is inseparable from the pathological features of AD, especially the deposition of Aβ, and is involved in the pathogenesis of AD.
Some researchers have shown that production of polyphenol-derived phenolic acids by intestinal flora attenuated Aβ oligomerization in AD [12]. Another study reported that targeted photobiomodulation therapy with light at 630 nm, 730 nm, and 850 nm could reverse Aβ-induced imbalance of the gut flora in AD mice, thus improving the learning ability, amyloid plaque deposition, tau phosphorylation, and microglial cell inflammation of the modeled mice [13]. All these findings suggest that change in the gut flora may be associated with the pathogenesis of AD through multiple pathways and channels, and targeting therapy via the gut flora may be a promising treatment for AD.
INTESTINAL MICROORGANISMS
The intestine is a complex and active micro-ecosystem [14]. Inhabited by a diverse population and a large number of microorganisms, known as the second largest gene pool of the human body, the intestinal flora has a complex symbiotic relationship with the host [15]. Based on the 16sRNA sequencing results [16], more than 1000 species of bacteria have been detected in the human intestine. Almost 99% human intestinal strains are anaerobic, with the most common specialized anaerobic bacteria and bacillus thuringiensis, accounting for about 51% and 48% respectively, leaving only about 1% specialized aerobic, parthenogenic and microaerobic bacteria [17]. Intestinal microorganisms can help the body digest and absorb food, prevent the invasion of pathogenic microorganisms, and regulate the growth, development and reproduction of intestinal mucosal cells [18]. They play important roles in maintaining the normal function of the human body.
GUT MICROBIOTA IN AD PATIENTS
The composition of human intestinal flora is not invariable and could be affected by multiple factors such as dietary habits [19], living environments [20], and illnesses [21]. Studies have demonstrated that the structure of the gut flora can differ between healthy people and AD patients [22, 23]. One of the main differences is the change in diversity, uniformity, and abundance of the gut flora. The gut flora species in healthy people are rich in Firmicutes, Actinobacteria, Bacteroidetes, and Verrucomicrobia [24], whereas, known intestinal microorganisms such as Actinobacteria, Aspergillus, Escherichia coli, Phyllobacterium, Aspergillus, Phyllobacterium, and Treponema play a key role in the pathogenesis and progression of AD [25]. The actinobacterium is one of the main bacterial species in both healthy people and AD patients, suggesting that the balance of Actinobacteria in the body may play an important role in maintaining the normal operation of the body. However, no convincing evidence is currently available to evaluate its contribution to the organism. Some researchers measured the intestinal flora in 3-month-old non-transgenic mice (NoTg) and triple transgenic mice with AD (3xTg-AD), and found that actinomycetes and TM7 relative abundance were decreased in 3xTg-AD mice [26]. Clinical research has also demonstrated that AD patients have less Eubacterium rectale and Bacteroides fragilis [27]. In addition, animal experiments have shown that Firmicutes and Bacteroidetes are the most predominant phylum in APP/PS1 mice [28]. There may be some differences in the results reported by different studies due to different means of detection. Currently, the most urgent issue is to clarify the role of these flora in AD and determine whether there is a competitive relationship between them and whether these gut flora species are also involved in pathways to participate in the development of the disease or accompanied by lesions in other parts of the body that jointly affect the composition of the flora.
PATHOGENESIS OF Aβ IN AD
The amyloid cascade hypothesis has long been thought of as one of the key pathogenic mechanisms of AD [29]. However, the exact mechanism of how β-like proteins mediate the onset of disease remains largely unknown. Aβ is produced by sequential cleavage of amyloid-β protein precursor (AβPP) by β-site amyloid precursor protein cleavage enzyme 1 (BACE-1) and γ-secretase [30]. Protein peptides formed during AβPP cleavage can be classified into Aβ1 - 40 and Aβ1 - 42 according to the number of amino acid residues. To our surprise, the cleavage of Aβ1 - 40 and Aβ1 - 42 by BACE1 results in the formation of a common Aβ34 intermediate, which is accompanied by elevated cerebrospinal fluid levels in patients in the early stages of the disease. In addition, researchers found that Aβ34, which is mainly distributed around brain capillaries near pericytes in AD patients, is time- and dose-dependent with Aβ40, and when the uptake of Aβ40 is reduced, the production of Aβ34 is also inhibited and the function of BACE1 suffers [31]. Although there is much research on Aβ as a whole, there is little research on Aβ43, an important component of amyloid plaques is Aβ43 has an aggregation tendency and is associated with neurotoxicity. At the same time, it was surprising to find by immunostaining that the release area of Aβ43 was significantly higher than that of Aβ40 [32]. Some studies suggested that Aβ43 may be a trigger for the aggregation of other monomers into aggregates of different conformations. By comparing the contribution of Aβ38, Aβ40, and Aβ43 aggregates to Aβ injected in the brain of mice, it was found that Aβ42 was not increased in neither Aβ38 nor Aβ40, while an increase in Aβ42 aggregates was detected in the brain of all mice inoculated with Aβ43 [33]. More interestingly, liquid mass spectrometry showed that Aβ43 was stimulated by Aβ48 in PS1 mutants, which, together with Aβ42, was involved in the formation of senile plaques, inducing the early onset of the disease, namely familial Alzheimer’s disease (FAD) [34]. Although the intestinal brain axis is known as a communication bridge between the intestinal microflora and Aβ, the size of its contribution to various subtypes of Aβ has not yet been defined, and there is not enough evidence to confirm the link between Aβ34/Aβ43 and other subtypes of Aβ, and the association between them needs to be further explored in future.
Monomers, oligomers, protofibrils, and insoluble fibrils are pumps for building Aβ [35]. Although all of them form part of Aβ, they are very different from each other and appear in different stages of AD and exert diverse targets of action. Soluble amyloid-β oligomer (AβOs) production and inflammatory activation are thought to be the main causes of early AD [36]. AβO impairs short-term object recognition memory and reduces glutamate uptake and oxidation in the hippocampus. In addition, AβOs reduce spare respiratory capacity, decrease ATP levels, impair cellular processing of Ca2 +, and cause mitochondrial swelling in hippocampal synaptosomes [37]. Synaptic damage and loss underlie the pathophysiology of AD, leading to reduced cognitive function [38]. Meanwhile, Aβ oligomers can be arranged in two patterns, forming neurotoxic oligomers and fibrous or nontoxic amorphous aggregates [39]. Although Aβ is known to influence the occurrence and progression of the disease, the mechanisms involved remain largely unknown.
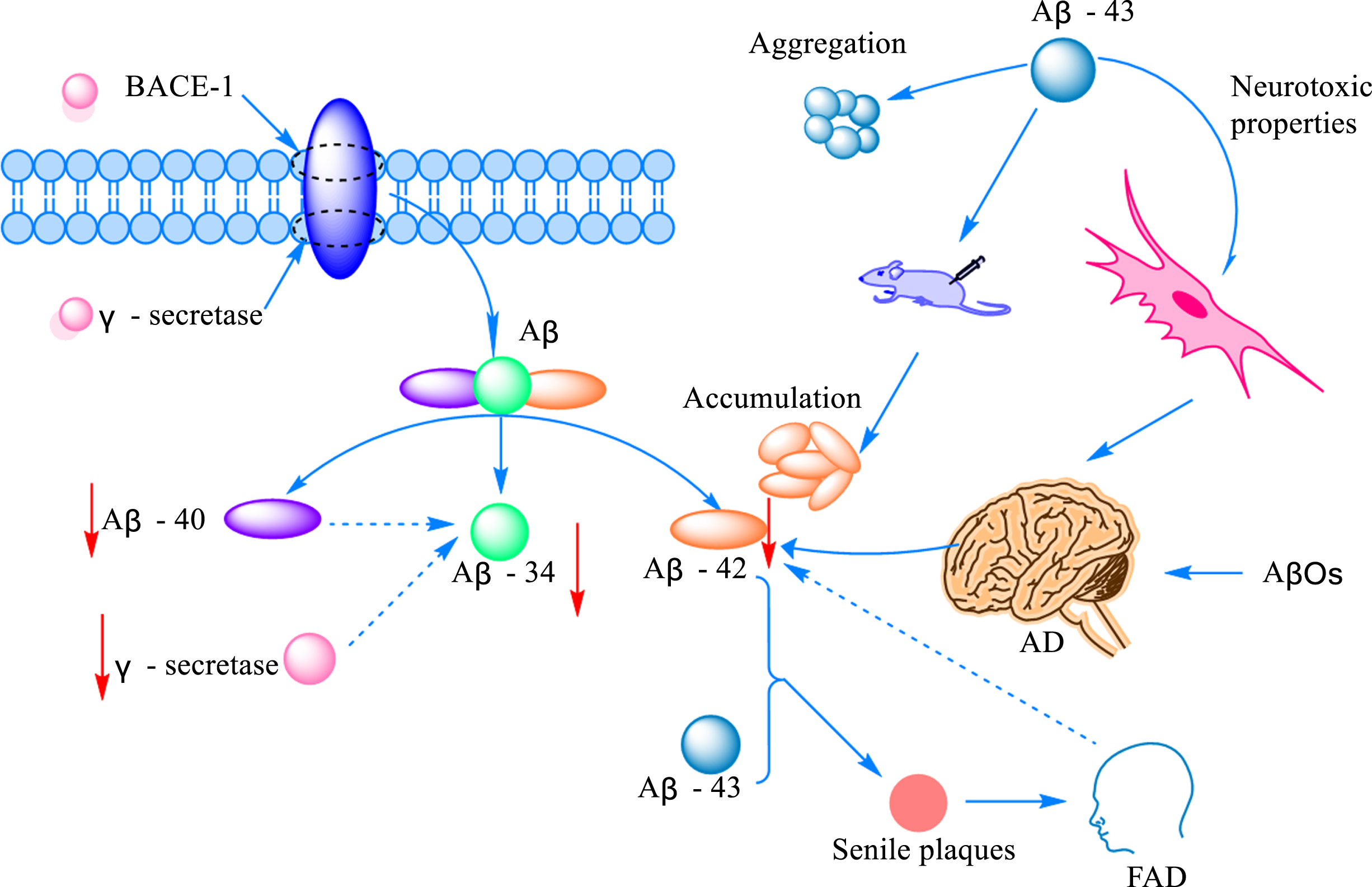
The mechanism of Aβ-mediated AD onset. The amount of Aβ produced by cleaved AβPP increased with the increased expression of protein amyloid pre-β-degrading enzyme 1 (BACE-1) and γ-secretase. Cleavage generates Aβ peptides of various lengths, commonly including Aβ40 and Aβ42, as well as the intermediate Aβ34. The production of Aβ34 correlates with the activities of Aβ42 and γ-secretase. Aβ43 is another Aβ protein peptide with obvious aggregation and neurotoxicity. Aβ42 can promote plaque formation and induce the occurrence of FAD at the same time, but studies have found that Aβ42 was decreased in the brain of AD and FAD patients.
NEW STRATEGIES FOR AD TREATMENT
It is discovered in recent years that peripheral immunity such as the gut flora plays an important role not only in AD not only by regulating the central immune system but through the brain-gut axis pathway. The gut flora also quietly participates in the clearance of Aβ through the cerebro-intestinal pathway, and this new understanding about the gut flora may shed new light on the treatment of AD patients.
Gut flora as the behind-the-scenes regulator of Aβ
The gut flora has become a hotpot of research in recent years. Many interesting things have also been discovered by linking the gut flora to the pathogenesis of neurodegenerative diseases through the brain-gut axis. It has been reported that increased intestinal permeability and blood-brain barrier caused by microbiota dysbiosis may influence the pathogenesis of AD. Inflammatory microbes in the gut flora are associated with peripheral inflammation and brain Aβ deposition in individuals with cognitive impairment [40]. Even intestinal microbial metabolites, such as pro-inflammatory factors, short-chain fatty acids (SCFAs), trimethylamine N-oxide, and neurotransmitters, can have an impact on the development of AD [41, 42]. Metabolic dysfunction of the gut flora accelerates the progression of AD as represented by increased Aβ retention, neuroinflammation, and loss of the cognitive ability [43]. Therefore, the role of the gut flora in the pathogenesis of AD cannot be ignored.
It was found that quercetin-3-O-glucosinolate (Q3G), a pharmacologically active flavonol glucosinolate, could alleviate brain insulin resistance (IR) by modulating the brain-gut axis to restore Aβ1 - 42-induced SCFA reduction leading to cognitive dysfunction, and correct gut flora dysbiosis [44]. High doses of lifelong antibiotics (ABX) administered to APPPS1-21 mice could reduce Aβ amyloidosis. In addition, transplantation of fecal microbiota transplants from transgenic (Tg) or WT male donors into ABX-treated male mice could completely restore Aβ amyloidosis, plaque-localized microglia morphology, and Aβ-related degenerative changes [45]. Lactobacillus plantarum C29 and fermented defatted soybeans (DW2009) significantly inhibited Aβ and β/γ-secretase activity, as well as blood and fecal lipopolysaccharide levels and enterobacteriaceae flora, and increased lactobacillus/bifidobacteria flora, thereby reducing cognitive dysfunction in 5XFAD Tg mice [46]. Phenyl-γ-valerolactone as the main gut microbiota-derived metabolite of flavan-3-ols could reduce the amount of intracellular and extracellular Aβ1 - 42 peptides by modulating neuronal cellular protein hydrolysis [47]. Some researchers also found that SCFA was effective in inhibiting Aβ aggregation in vitro [48]. Akkermansia muciniphila (Akk), the only representative human intestinal Verrucomicrobia genus, was found to reduce the Aβ40 - 42 levels in the cerebral cortex of APP/PS1 mice and improve the correct rate of the Y-maze test [49]. The probiotic Clostridium butyricum could prevent cognitive impairment, Aβ deposition, microglia activation, and inflammatory factor production in the brains of APP/PS1 mice, and treatment with its metabolite butyrate also reduced CD11b and COX-2 levels and decreased Aβ-induced NF-κB p65 phosphorylation in microglia [50]. In addition, the gut flora composition in Aβ1 - 42-induced AD mice was greatly altered, especially in some beneficial and inflammation-associated bacteria, such as Lactobacillus, Bifidobacterium, Desulfovibrio, Treponema, Mucor, Alistipes, Anaerotruncus, and Rikenella [51]. However, some other studies also reported that although probiotic supplementation significantly reduced intestinal inflammation and intestinal permeability, it had a minimal effect on Aβ, cytokine, or glial proliferation in the brain [52]. More experiments are required to confirm whether probiotic therapy really helps remove Aβ, and more sensitive, accurate and reliable detection methods are needed to track the whole process of Aβ from formation to removal.
Another interesting finding is that the composition of the intestinal flora and the level of Aβ are surprisingly correlated with gender. A study [53] analyzed sex differences in the effect of oral probiotics in the AppNL - G - F mouse AD model and found that there was a significant association between the bacterial genus and the Aβ plaque load, gliosis and memory in AppNL - G - F mice. In addition, no significant improvement in Aβ plaque formation, microglial cell proliferation and brain TNF-α and memory was observed between female AppNL - G - F mice fed with probiotics and corresponding normal controls, whereas a moderate innate pro-inflammatory response to the probiotic treatment and an adaptive response to the antibiotic treatment were observed in male AppNL - G - F mice. In addition, specific gut microbiota changes were found to be associated with Aβ level and affect women more pronouncedly than men [54]. There is ample evidence that estrogen level has a close correlation with cognitive function in AD [55, 56]. However, long-term estrogen use increases the risk of breast cancer and some other diseases, and may exacerbate oxidative stress and inflammatory response [57, 58]. In addition, the recommendations for AD prevention in evidence-based prevention of AD published in 2020 clearly state that estrogen replacement therapy is not recommended. Therefore, whether intestinal flora can inversely regulate estrogen secretion levels and replace estrogen as an alternative therapy for AD needs to be further verified [59].
Effect of gut flora dysbiosis on Aβ
The main manifestation of persistent imbalance in the gut flora, also known as intestinal dysbiosis [60], is the structural change of the microbial community from specialized anaerobic bacteria to parthenogenic anaerobic bacteria [61]. In recent years, an increasing number of studies have demonstrated that intestinal dysbiosis contributes to the pathogenesis of AD by promoting disease progression through excessive consumption or production of metabolic derivatives. A previous study [5] observed dysregulated intestinal homeostasis prior to brain Aβ accumulation in Tg2576 mice. In addition, intestinal microecological dysregulation led to the secretion of amyloid and lipopolysaccharide and increased the permeability of the intestinal and blood-brain barriers [62]. It also promoted the formation of β-fold conformation of the protein into toxic Aβ, which promoted loss of synaptic connections, cellular dysfunction and neurodegenerative lesions [63]. In fact, Aβ is considered to be an antimicrobial peptide with antimicrobial properties, involved in the innate immune response. However, Aβ exhibits deleterious properties in a state of dysbiosis of the gut flora. Bacterial amyloid acts through molecular mimicry, hence misfolding and cross-seeding Aβ peptides, and inducing the activation of microglia [64]. It has been shown that phosphorylation of tau proteins and acceleration of protein amyloid deposition are both mediated by 5-lipoxygenase, which is strongly associated with dysbiosis of the gut flora [65]. Dodiya et al. [66] found that ABX cocktail treatment changed the gut microbial composition and reduced Aβ deposition in two independent transgenic strains (called APPSWE/PS1ΔE9 and APPPS1-21) of male mice, indicating that dysbiosis of the intestinal flora plays a key role in mediating the formation and deposition of Aβ, and suggesting that a balanced gut flora and rational use of antibiotics may play a role in improving AD.
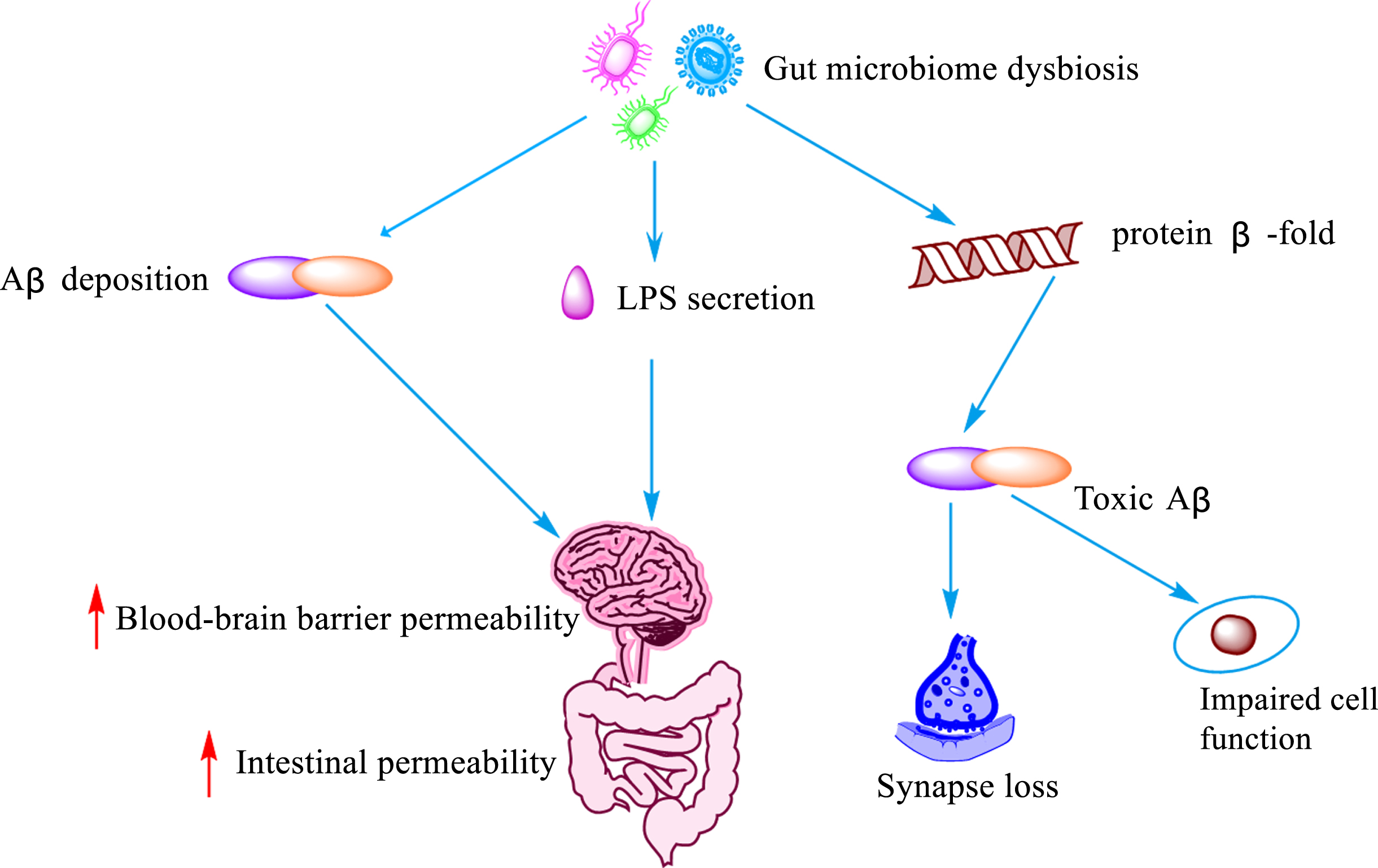
Influence of intestinal flora imbalance on Aβ. Dysregulation of the gut microbiota leads to the deposition of Aβ, LPS secretion, and misfolding of protein β-fold, resulting in increased blood-brain barrier and gut permeability, cellular impairment and loss of synaptic function lose.
Intestinal dysbiosis induces IR to exacerbate Aβ deposition
Type 2 diabetes mellitus (T2DM) is an important risk factor for the development and progression of AD [67]. Earlier studies found a positive correlation between insulin and beta-40 and beta-42 by monitoring biochemical markers in healthy people, diabetic patients, AD patients, and diabetic AD patients [68]. Insulin resistance (IR) is inextricably linked to the development and progression of AD and is considered as one of the essential pathogenic mechanisms and features of AD [69, 70]. It is reported that IR promotes the formation and deposition of Aβ [69, 71]. Iwatsubo et al. [72] explored the relationship between IR and amyloid by constructing an insulin-resistant AD mouse model and found that insulin deficiency in the brain decreased Aβ clearance and increased Aβ deposition. They also found that the static and dynamic incubation of insulin with the Aβ isoform Aβ42 produced a serious effect on Aβ42 protofibril formation, which interfered with Aβ42 self-assembly and prevented the formation of soluble oligomers [73]. Furthermore, their study unexpectedly found that glucagon-like peptide-1 analog liraglutide, an antidiabetic drug, reversed the phosphorylation status of insulin receptor and insulin receptor downstream signaling molecules insulin receptor substrate 1, protein kinase B, and glycogen synthase kinase 3 beta, improved peripheral IR, and reduced BACE-1 enzyme activity and Aβ formation in insulin-resistant cells [74]. However, with the in-depth investigation of the gut flora in recent years, a strong link between intestinal flora and IR has been disclosed. Diabetic mice were found to have severely dysregulated intestinal flora in the early days of life, and treatment with the insulin receptor antagonist S961 increased the intestinal permeability and disrupted the integrity of the intestinal barrier [75]. In addition, microbial diversity was reduced in IR iNOS–/– mice, while the abundance of Haemophilus and Bifidobacterium and Gram-positive bacteria was increased, which was accompanied with alteration in serum metabolites and metabolic dysregulation [76]. Some researchers found that there was a correlation between IR, gut flora and Aβ formation or deposition. They used Tetragonia Tetragonioides Kuntze (TTk) 70% ethanol extract to treat Aβ (25–35) infused rats and found that it could enhance the memory function, improve hippocampal insulin signaling, alleviate IR, prevent protein amyloid deposition, and improve intestinal microbiology [77]. However, the mechanisms involved are not clear and need to be further explored.
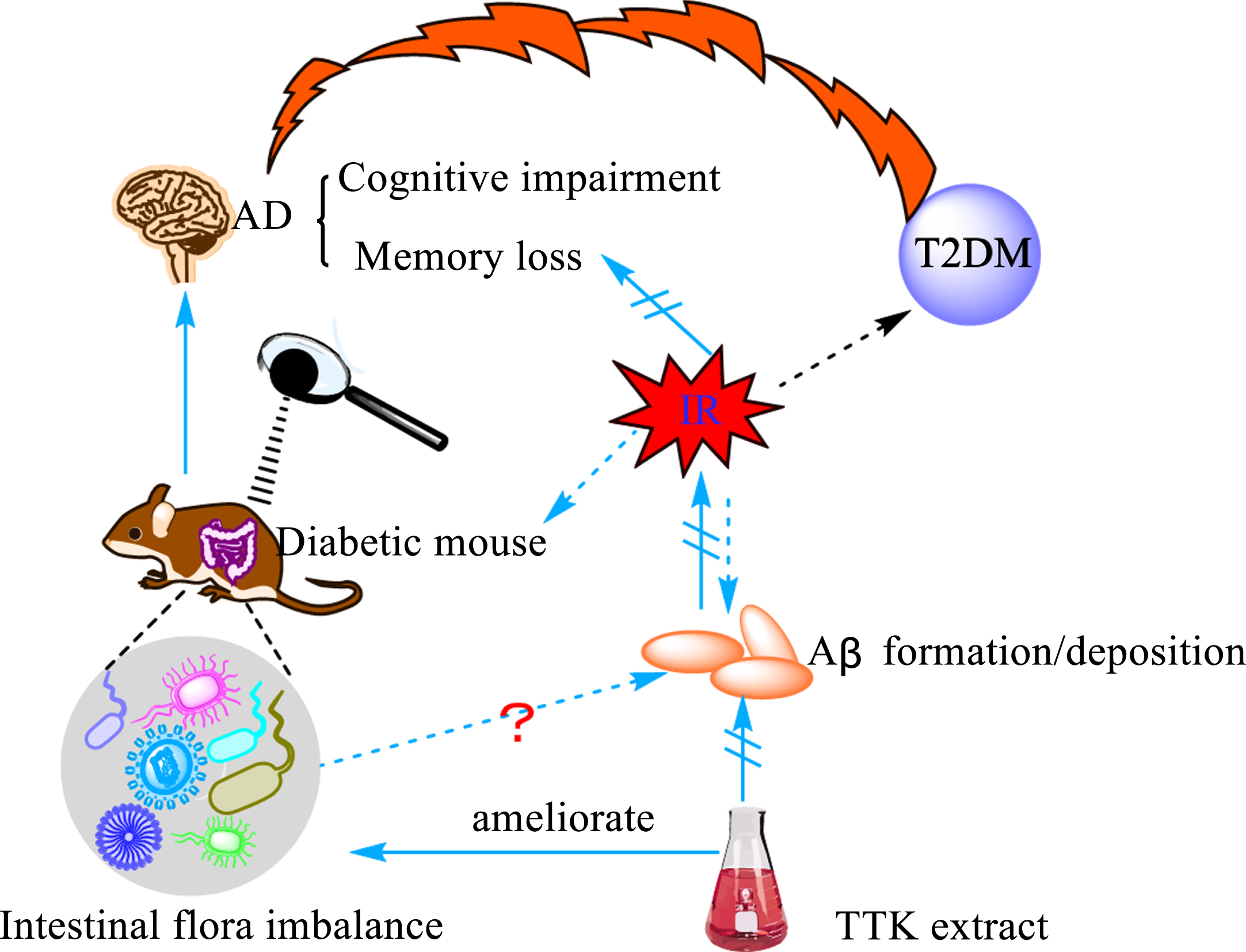
The bridge between Aβ and intestinal flora. Insulin resistance is a significant feature of type 2 diabetes (T2DM) and one of the main mechanisms underlying the occurrence of diabetes. The study found that diabetic mice developed severe intestinal microbiota disorders in the early stages of the disease, accompanied by cognitive dysfunction and memory loss. Studies have shown that insulin resistance (IR) promotes the deposition of Aβ. Under the action of Tetragonia Tetragonioides Kuntze (TTK) alcohol extract, the phenomenon of intestinal dysbiosis, the formation and deposition of Aβ and the symptoms of cognitive impairment and memory loss were all alleviated.
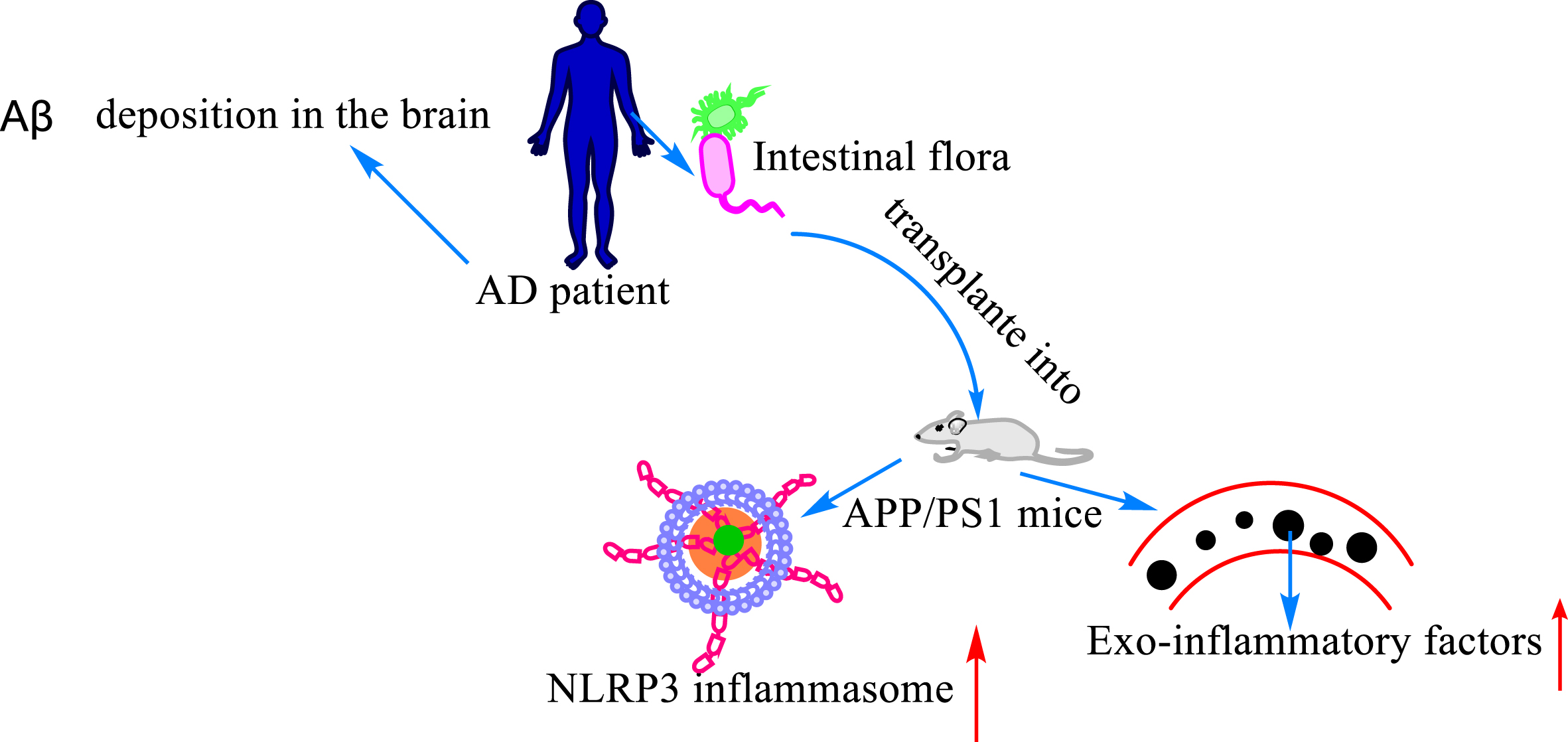
Relationship between Aβ, gut flora, and neuroinflammation. Deposition of Aβ in the brain is one of the hallmarks of AD. The expression level of inflammasome and inflammatory factors was increased in the peripheral blood after transplanting the intestinal flora of AD patients into APP/PS1 mice.
Gut flora dysbiosis leads to neuroinflammation and exacerbates Aβ accumulation
It is well established that neuroinflammation can induce the onset of AD. Although great efforts have been made to disclose the mechanism that mediates AD onset, no definitive answer has been found. As the gut flora has become a popular topic of research, it is surprising to find that there is also a link between the gut flora of AD patients and the development of neuroinflammation. Some researchers transplanted gut flora from AD patients into APP/PS1 mice, and found that the expression level of inflammatory vesicle NLRP3 was increased in the intestine of the mice accompanied with an elevated expression of exo-inflammatory factors in the peripheral blood. Congruously, mice receiving the gut flora from AD patients had more severe cognitive deficits than those not receiving the gut flora, accompanied with activation of microglia in the central hippocampus and increased expression level of neuroinflammatory factors [78]. Song et al. [79] studied the effect of tea polyphenols on the gut flora in mediating neuroinflammation and improving memory function in rats by D-galactose injection to construct a model of menopausal aging, and found that the model rats exhibited severe gut flora dysbiosis, increased permeability of the intestinal epithelium, increased expression of inflammatory proteins such as TLR4/NF-κB in the rat hippocampus, and excessive activation of microglia, which exacerbated the progression of neuroinflammation in the brain. However, there is a feedback effect between neuroinflammation and Aβ. AD is a disease characterized by amyloid deposits with neuroinflammation [80]. When neuroinflammation occurs, microglia are activated as immune cells of the nervous system. Activation of microglial cells leads to amyloid overload, which further amplifies the pro-inflammatory response [81].
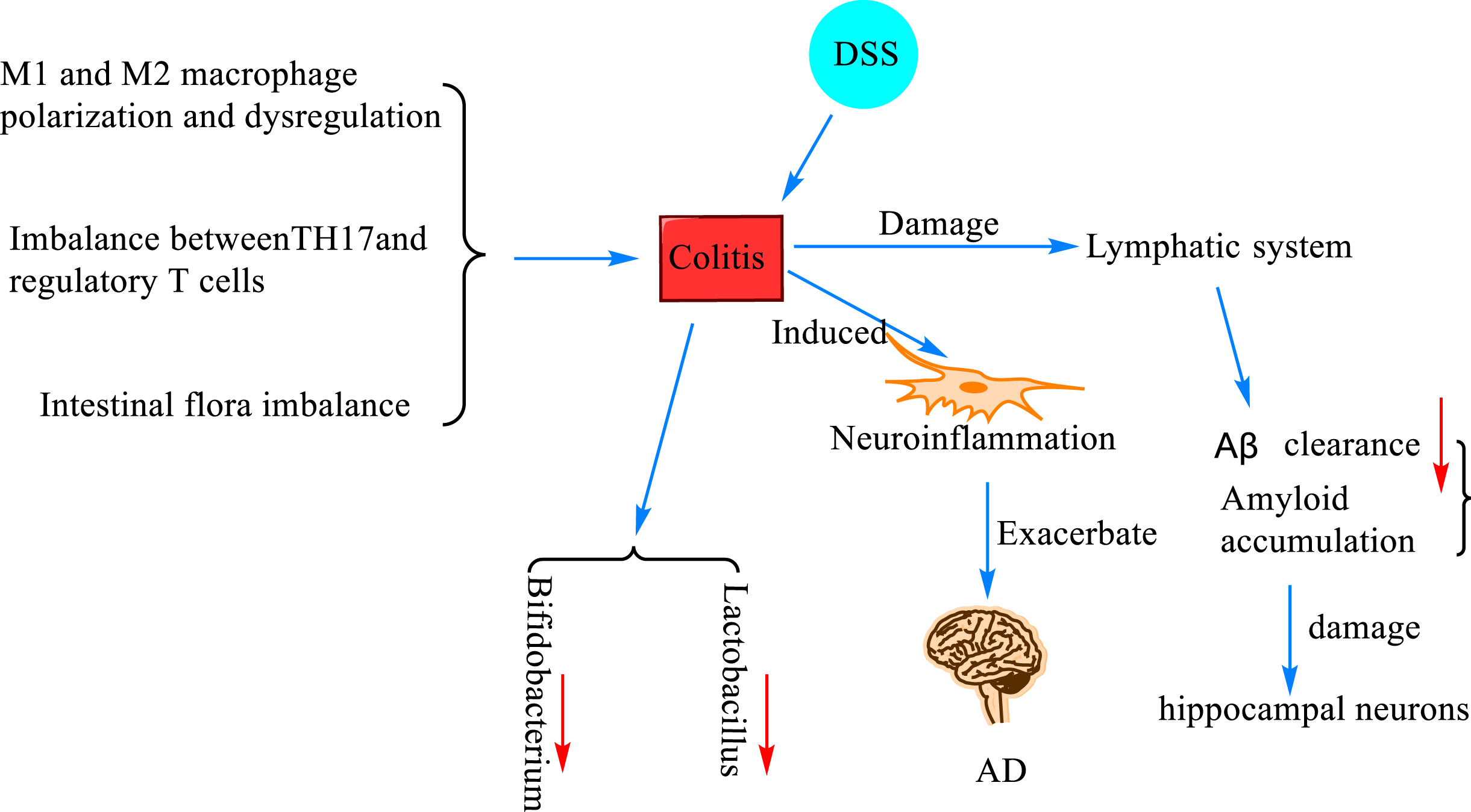
The mechanism of colitis and Aβ deposition. Colitis was induced when M1 and M2 macrophages were polarized and dysregulated causing imbalance between T helper 17 (TH17) and regulatory T cells (Treg cells) and intestinal flora dysbiosis. In mice with dextran sodium sulfate (DSS)-induced ulcerative colitis, the number of Bacillus fragilis and Escherichia coli was increased, and the number of Bifidobacteria and Lactobacilli was decreased, and neuroinflammation was induced, resulting in damage to the lymphatic system and decreased Aβ clearance, which led to the accumulation of amyloid and damage of hippocampal neurons, which further promotes the progression of AD.
Intestinal dysbiosis-induced colitis exacerbates Aβ accumulation
Colitis, in layman’s terms, is an inflammatory lesion of the colon caused by various causes. Colitis can develop when M1 and M2 macrophages are polarized and dysregulated [82], when there is an imbalance between T helper 17 (TH17) and regulatory T cells (Treg cells) [83], and when the gut flora is dysregulated [84]. A study [85] found that the number of Bacillus fragilis and Escherichia coli was increased, and the number of Bifidobacterium and Lactobacillus was decreased in mice with ulcerative colitis induced by dextran sodium sulfate (DSS) modeling. They also reported that colitis exacerbated the progression of AD by inducing neuroinflammation, which may be attributed to damage to the lymphatic system responsible for clearing Aβ. Later, researchers constructed a DSS-induced inflammatory bowel disease (IBD) model and found that DSS-induced colitis impaired the spatial memory of the modeled mice, accompanied with activation of microglia and an increase in A1-type astrocytes. It also impaired the lymphatic system responsible for clearing Aβ, leading to the accumulation of amyloid and resulting in the loss of hippocampal neurons [86]. In addition, Sohrabi et al. [87] created an IBD model by feeding DSS to C57BL/6 wild mice and AppNL - g - f mice, and found by brain histological and biochemical assessment that more insoluble amyloid plaques were formed in DSS-treated AppNL - g - f mice, and CD68 immunoreactivity in microglia trended to decrease. These findings further validate the previous suspicion that there is indeed a strong relationship between colitis and neuroinflammation and Aβ.
CONCLUSIONS AND FUTURE PERSPECTIVES
Based on a large body of research on the brain-gut axis, this review aims to elucidate the link between AD and the gut microbiota and AD biomarker Aβ to provide evidence to support clinical research efforts. A large number of in vivo experiments have shown that intestinal flora imbalance aggravates the accumulation and deposition of Aβ, and both the intestinal flora and intestinal metabolites play an important role in the regulation of Aβ. Given the minimal adverse reactions of probiotics therapy in clinical application and the increased acceptance of people on probiotics, the use of probiotics to regulate intestinal flora for the treatment of diseases has gradually become the mainstream. Clinical researchers have tried to use probiotics therapy to observe whether improving intestinal flora has a positive therapeutic effect on non-alcoholic fatty liver disease. The results demonstrated that probiotics therapy can improve liver enzyme levels in patients with nonalcoholic fatty liver disease, regulate lipid metabolism, improve IR, and reduce inflammation levels, showing a good therapeutic effect [88]. Other studies have shown that probiotics have a positive effect on improving diabetic nephropathy. After taking probiotics, various renal function indicators were corrected, lipid metabolism and insulin resistance were improved, and inflammation and oxidative stress levels were reduced [89]. And so far, the novel coronary pneumonia epidemic (COVID-19) has been plaguing people all over the world. Administration of mixed probiotics significantly reduced the inflammatory markers in the body of patients with interstitial pneumonia [90]. It seems that for many intractable diseases, probiotic therapy has become an alternative therapy when no suitable drug or method has been found. Probiotic therapy can correct various indicators of the body and bring about good results, bringing hope for treatment to both patients and clinicians. Therefore, in the future, treating AD by using probiotics to modulate the gut microbiota may become a strategic treatment approach.
As AD is a central nervous system disease of the brain, its connection with intestinal flora cannot be separated from the subphrenic vagus nerve. The subphrenic vagus nerve is thought to be a major regulatory pathway between the brain and gut microbes, which modulates the structural composition of the gut microbiota [91]. It was found that vagotomy had a detrimental effect on the normal gut microbiome composition in healthy people [92]. A study [93] showed that atrial natriuretic peptide could effectively alleviate LPS-induced hippocampal inflammation and cognitive impairment through the subphrenic vagus nerve-mediated gut-brain axis. Another recent study [94] found that when Aβ or tau fibrillin and brain lysates from AD patients were injected into 3xTg mice, these signals traveled from the gut to the brain via the vagus nerve, causing AD-like lesions and cognition disfunction. Using vagotomy can selectively attenuate this signal, alleviate Aβ and tau pathology, and restore learning and memory function in mice. Therefore, when we consider the relationship between the gut-brain axis in regulating AD disease and the gut microbiota, and the relationship between the gut microbiota and Aβ, the vagus nerve should not be missed. Comprehensive consideration of all these factors may better help unravel the confusion.
The main purpose of this article is to provide a theoretical basis for elucidating the role of gut microbiota and gut microbial metabolites in AD treatment by analyzing the relationship between gut microbial composition and Aβ and the mechanism of action of Aβ in AD.
Footnotes
ACKNOWLEDGMENTS
Special thanks to the National Natural Science Foundation of China (81860640) for funding my project. I would also like to thank my co-authors for helping me search the literature and fill in the gaps in the article, and my corresponding authors for helping me revise and respond to the manuscript in a timely manner. In addition, I am also very grateful to the reviewers for giving me many valuable comments, and also thank the editors for their recognition of my article. Because of your encouragement and support, I have successfully reached the standards of the Journal of Alzheimer’s Disease.