
Abstract
Given the acknowledged lack of success in Alzheimer’s disease (AD) drug development over the past two decades, the objective of this review was to derive key insights from the myriad failures to inform future drug development. A systematic and exhaustive review was performed on all failed AD compounds for dementia (interventional phase II and III clinical trials from ClinicalTrials.gov) from 2004 to the present. Starting with the initial ∼2,700 AD clinical trials, ∼550 trials met our initial criteria, from which 98 unique phase II and III compounds with various mechanisms of action met our criteria of a failed compound. The two recent reported phase III successes of aducanumab and oligomannate are very encouraging; however, we are awaiting real-world validation of their effectiveness. These two successes against the 98 failures gives a 2.0% phase II and III success rate since 2003, when the previous novel compound was approved. Potential contributing methodological factors for the clinical trial failures were categorized into 1) insufficient evidence to initiate the pivotal trials, and 2) pivotal trial design shortcomings. Our evaluation found that rational drug development principles were not always followed for AD therapeutics development, and the question remains whether some of the failed compounds may have shown efficacy if the principles were better adhered to. Several recommendations are made for future AD therapeutic development. The whole database of the 98 failed compounds is presented in the Supplementary Material.
INTRODUCTION
Alzheimer’s disease (AD) is a chronic debilitating neurodegenerative disorder characterized by the onset of cognitive impairment, progressing to dementia and culminating in premature death. It is the most common cause of dementia, accounting for up to 75% of all cases [1], and surpasses other types such as vascular, frontotemporal, Lewy body, mixed, etc. Other salient features of AD include behavioral changes (e.g., depression, agitation, aggression), sleep disturbances, and loss of bodily functions. This is a particularly nefarious affliction that robs its victims of the memories and self-knowledge that define them as a sentient being. The sporadic or late-onset variants account for 94–99% of all AD patients, with the early-onset variant accounting for the remainder [2, 3]. As many as four distinct late-onset variants have been identified [4], and such heterogeneity is thought to reflect differing and possibly concurrent pathways leading to the neurodegeneration and disease progression. The underlying pathophysiology of AD is extremely complex and as yet not fully understood. The hallmark pathological brain changes include an accumulation of amyloid-β (Aβ) plaques arising mainly from the Aβ42 variant, and formation of tau neurofibrillary tangles. These are associated with the neuronal loss and neurodegeneration characteristic of AD [3, 5]. Various physiologic pathways are thought to play major roles in the progression of the disease, such as apolipoprotein E (APOE)-mediated cholesterol transportation and metabolism, neuroinflammatory response, energy utilization, and vascular burden [6–11].
Today in the US alone, an estimated 5.8 million people suffer from AD, afflicting 10% of people above 65 years of age [1]. This number will grow due to the rapid aging population, with projections of almost 14 million affected individuals by 2050. This will add tremendous burden to the already unsustainable cost of the US healthcare system. Currently, an estimated $240 billion USD is spent annually on healthcare, long-term care, and hospice services for people aged ≥65 years with dementia [1]. Additionally, family members and other unpaid caregivers provide services estimated at over $230 billion USD annually.
Current validated AD treatments modestly slow its progression, and none can reverse it. Although two recent developments offer hope, real-world validation of their effectiveness are not yet available. In 2021, Biogen/Eisai’s aducanumab (Aduhelm), a monoclonal antibody designed to bind and eliminate aggregated Aβ plaques was approved. Initially, the interim analysis failed to show efficacy, but upon analyses of a more complete dataset, efficacy was reported in a subset of patients at the highest dose who received all infusions [12]. The Food and Drug Administration (FDA) approved this drug despite the near unanimous recommendation by the scientific advisory committee to reject approval, and was based on the surrogate endpoint of brain Aβ plaque reduction; and with the requirement to run a post-marketing trial [13]. More recently, the European Medicines Agency rejected approval, and Japan’s Pharmaceuticals and Medical Devices agency is continuing deliberations and has requested additional data [14, 15]. In late 2019, a seaweed-derived oligosaccharide called oligomannate (GV-971), that purportedly works through intestinal microbiomes and anti-inflammatory actions (and may also impact amyloid and tau), was conditionally approved by China’s National Medical Products Administration for mild-to-moderate AD patients [16]. Evidence for approval was based on a single phase III trial of Chinese patients, and the sponsor (Green Valley) has since initiated a global phase III trial. The five earlier approved drugs (one was subsequently removed from the market) indicated for AD are symptomatic treatments that are modestly effective. These treatments are a N-methyl-D-aspartate glutamate receptor antagonist and three acetylcholinesterase inhibitors [3]. The last of these were approved by the FDA and European Medicines Agency in 2003, although a fixed-dose combination of two previously approved drugs was approved in 2014.
The lack of success in AD drug development compels us to ask: What is wrong with our current approach? Is the failure rate due to faulty drug development processes, lack of understanding of the underlying AD pathophysiology such that we are incorrectly pursuing biologic targets, or perhaps a combination thereof? To gain some insights, we reviewed AD clinical trials (interventional phase II and III) from 2004 to the present and investigated potential reasons for the failures. Thus, the overall goal of this review was to better understand the reasons for the myriad clinical trial failures, in order to provide insights and recommendations of how to improve the success rate of future clinical trials. Other researchers have documented potential reasons for AD clinical trial failures [6, 17–19]. However, this is the first to our knowledge to systemically and exhaustively examine all clinical trials failures (from the ClinicalTrials.gov database) going back to 2004 to the present, to characterize the failed compounds, and to uncover and classify the potential reasons for the lack of success.
APPROACH TO EVALUATION
We describe here: 1) methods used to search and select the clinical trials for analysis; 2) commonly used cognitive/functional/global assessment batteries and AD biomarkers used in these clinical trials; 3) putative mechanism of action (MOA) of the compounds that were pursued; and 4) limitations of our approach and evaluation methods.
Compound and clinical trial selection criteria
We performed an exhaustive review of AD com-pound and clinical trial failures occurring between 2004 to 2021. Our main information source was ClinicalTrials.gov (clinical trial database), supplemented with PubMed (scientific publications), Alzforum (Alzheimer Research Forum, AD data-base), AdisInsight (pharmaceutical drug database), company web sites, and news releases. The inclusion and exclusion rules applied to compounds and clinical trials for selection into the evaluation are listed in Table 1. Phase II and III compounds were deemed failures if the trial efficacy endpoint was not met, significant adverse events occurred to prevent continuation of the program, or there were reports that the program was discontinued.
Inclusion and exclusion rules applied to compounds and clinical trials for selection into the evaluation
Tests and measurements
Key aspects of our evaluation involved AD drug efficacy tests and AD biomarkers.
AD clinical trials measured drug efficacy using tests of cognitive abilities, functional performance, and/or global assessments, which serve as primary clinical endpoints. The following are the most commonly used tests in the failed clinical trials evaluated (this is not a full list of the available tests): Alzheimer’s Disease Assessment Scale - cognitive subscale (ADAS-Cog) –for cognitive impairment, the most commonly used primary clinical endpoint in past clinical trials; Clinical Dementia Rating scale - sum of boxes (CDR-SB) –for cognitive and functional performance; Alzheimer’s Disease Cooperative Study - Activities of Daily Living inventory (ADCS-ADL) –for daily living competency; Mini-Mental State Examination (MMSE) –for cognitive impairment; Clinician’s Interview-Based Impression Change - plus (CIBIC+) –for global assessment of change; Neuropsychological Test Battery (NTB) –for memory and executive function.
AD biomarkers help diagnose the disease and measure disease progression and treatment effectiveness. The following are the most commonly used AD biomarker tests [20, 21] from the failed clinical trials (this is not a full list of available tests): Positron emission tomography (PET) –for brain glucose metabolism, Aβ plaques, and tau (including phosphorylated-tau) brain deposits; Cerebrospinal fluid (CSF) –for Aβ (includes its variants such as 40 and 42), total tau and phosphorylated-tau levels, and neurofilament light; Blood –for neurofilament light; Genetic –APOE variants (i.e., ɛ4), and other genes; Magnetic resonance imaging –for volume changes in specific brain regions (e.g., hippocampus, ventricles) or whole brain.
Putative MOA class of compounds
The primary MOA of the failed compounds in our dataset are categorized in Table 2. In some cases, the MOA is not well understood or multiple potential mechanisms were listed. We assigned to each compound a “primary” putative MOA based on the weight of the evidence. Sources for this information included ClinialTrials.gov, Alzforum, and journal articles. The MOA classification categories and scheme we use has been adopted from other previous researchers [22–24].
Primary putative MOA class of AD compounds that failed in phase II or III, classified as disease-modifying versus symptomatic, and direct amyloid targets versus targets beyond amyloid
A distinction was made between compounds that are disease-modifying versus symptomatic treatments. This is an imperfect distinction as it is not always clear and some compounds may have both properties, but this framework was useful to categorize the numerous compounds for the purposes of the analysis. We assigned the primary classification based on the evidence; symptomatic compounds predominantly acted via neurotransmitter systems.
Distinctions were also made between compounds that primarily impact Aβ directly versus other primary mechanisms. Despite this delineation, compounds working via other MOAs may still impact Aβ as a downstream or indirect consequence of the drug action. We have therefore categorized the compounds’ MOA class as follows: Direct amyloid targets. Directly targets Aβ at one or more points along its biological pathway, from its synthesis, cleavage, post-transla-tional modification, aggregation, and its removal (either in soluble or aggregated form). Targets beyond amyloid. Targets many other known pathological processes involved in AD. The ones discussed in Table 2 are most represented in the clinical trials we assessed. Despite the discrete categorization framework that was adopted, there were overlaps in some of the underlying mechanisms.
Limitations of our approach and evaluation methods
Several limitations of our process should be acknowledged. First, this was a retrospective evaluation of the past clinical trials, without discussions of the promising treatments currently in development. Second, the database of clinical trials used in our evaluation was from ClinicalTrials.gov, with exclusion of other databases such as those in Europe and China. However, ClinicalTrials.gov is acknowledged to be the most comprehensive and rich database available and captures the vast majority of all trials in AD. Third, we did not directly access the failed clinical trial data in detail (where it was made publicly available) to examine any overall statistical trends, individual subject values, and correlations on multiple dependent measures (cognitive test results, biomarkers, etc.), various subgroups test results, etc. Such analyses would add value but was beyond the scope of our current review. And fourth, we did not evaluate the scientific strength of the drugs’ MOA and biologic targets; we merely classified them into the appropriate classes. A deeper analysis of the MOAs of these failed compounds to determine which could be most promising was beyond the scope of this current review.
OVERALL COMPOUND AND TRIAL EVALUATION RESULTS
As of November 30, 2021, the ClinicalTrials.gov database contained 2,695 clinical trials for AD. After applying filters of interventional phase II and III trials, removal of ongoing trials, and trial completion date between 2004 to the present, data from 543 trials were downloaded. Following additional criteria as outlined in Table 1, 98 unique compounds met our criteria as a failure. Figure 1 summarizes the results:
40 (41%) failed in phase III, and 58 (59%) failed in phase II; 63 (64%) were disease-modifying, and 35 (36%) were symptomatic; Of the 63 disease-modifying compounds, 32 (51%) failed in phase III, and 31 (49%) failed in phase II.
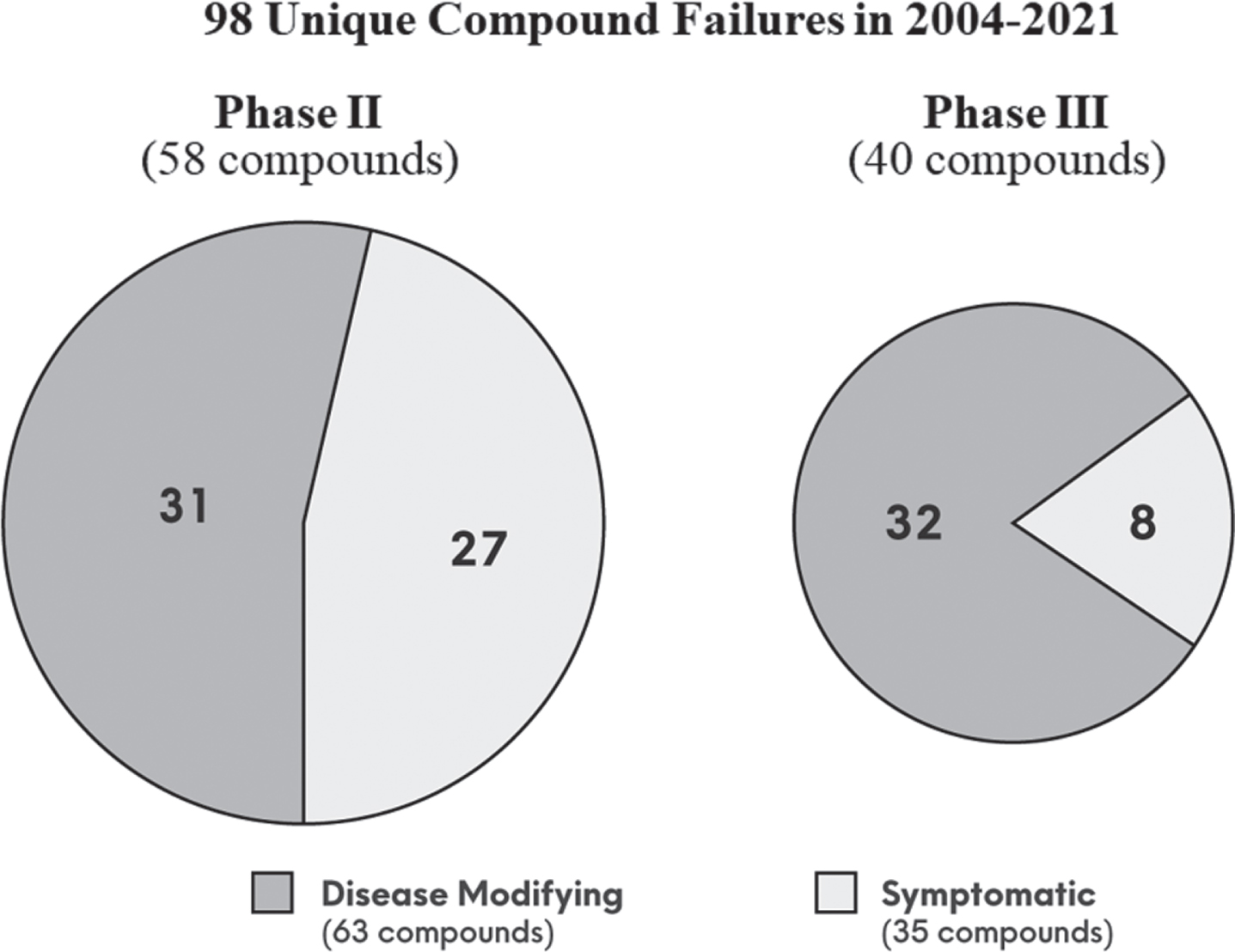
The 98 unique compound failures in clinical trial phase II and III, segmented by disease-modifying versus symptomatic, during the period of 2004 to 2021.
Of phase II compounds, there were another ∼50 where the development status was inactive or un-known without additional follow-up information, which did not satisfy our criteria for failure. Note that there is a tendency for biopharma sponsors to label phase II studies as “pivotal” (phase II/III or III) to attract greater investor attention; thus, the numbers of phase III versus phase II may possibly be inflated. Despite the lack of unanimous regulatory approval and lack of real-world evidence of effectiveness, aducanumab [13] and oligomannate [16] did clear the hurdle of phase III success. Thus, these two successes against the 98 failures, show a 2.0% AD drug development success rate for phase II and III, since the last novel approval in 2003.
Figure 2 provides a breakdown of the number of failed compounds that pursued the various MOA classes, and by phase of development. Amongst the disease-modifying compounds, the amyloid pathway at various levels was most targeted, accounting for almost a quarter of them (23 compounds or 23%). There were far fewer failed attempts pursing tau, the other core pathology of AD (
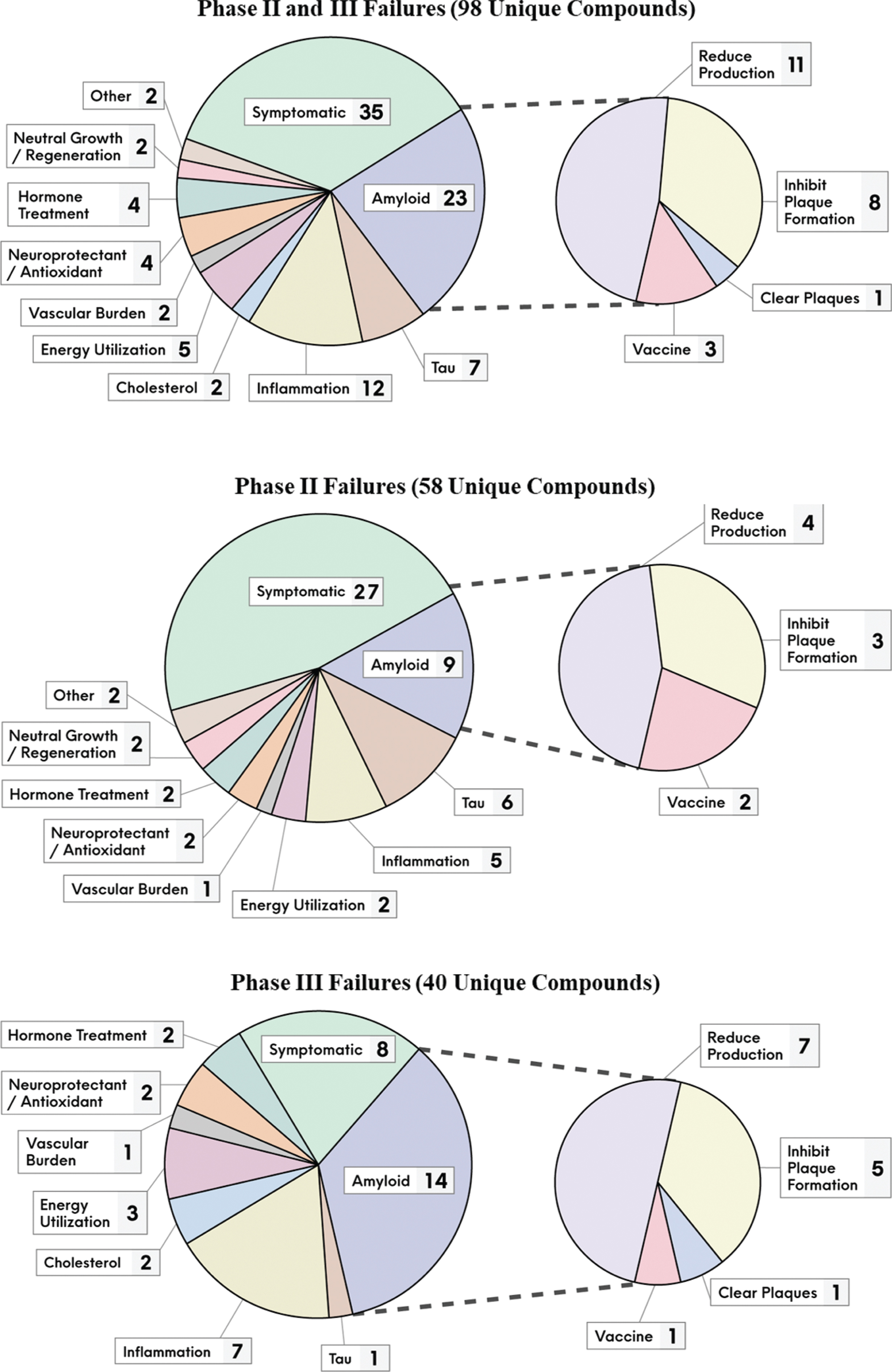
The unique failed compounds from 2004 to 2021, segmented by their putative MOA classes and development phase.
Since the start of 2018, 12 compounds failed in phase III, of which all were disease-modifying and nine targeted amyloid directly. The most recent failures were solanezumab [37] (inhibits amyloid plaque formation) and gantenerumab [38] (clears amyloid plaques) that targeted familial AD patients. This adds to the failures of solanezumab [39] and gantenerumab [40] with late onset AD patients. Several BACE inhibitor failures include elenbecestat [41], umibecestat [42], atabecestat [43], lanabecestat [44], and verubecestat [45, 46]. Other failures include crenezumab [47] which inhibits Aβ plaque formation (may also clear aggregated Aβ), amilomotide [48] an Aβ vaccine, azeliragon [49] which works via anti-inflammatory actions (and may also limit Aβ aggregation), nilvadipine [50] which decreases vascular burden, and pioglitazone [51] thought to work by increasing energy utilization. Refer to Supplementary Tables 1 and 2 for the complete list of the 98 failed compounds, along with their putative MOA and other relevant clinical trial information.
Evaluation of key drug development metrics (technical risk, development time, and development cost) quantitatively confirms the challenges of developing AD therapeutics (see Table 3). The historical industry average of AD versus all therapeutic categories shows the following: risk (measured by probability of success) is almost 9 times higher (2.0% versus 17.8%); time for development is almost 40% longer (7.6 years versus 5.5 years); and cost is over 2.2 times greater ($5.69 billion versus $2.56 billion).
Direct comparison of drug development risk (measured by probability of success), time and cost, for industry average of all therapeutic categories versus AD specifically. These comparisons were between datasets with parameters that were as similar as possible, including the time period of data collection
CLINICAL TRIAL FAILURE INSIGHTS
Our database was then used to perform bottom-up analyses to identify various potential reasons for the clinical failures. Although some factors were independently derived from examination of the dataset, they were confirmed from other authors who had previously made these observations [6, 17–19]. The evaluation showed that rational drug development principles were not always followed, and the factors can be categorized into 1) insufficient evidence for initiating the pivotal trials, and 2) pivotal trial design issues. We include supporting examples from the database for each factor.
Insufficient evidence for initiating pivotal trials
Ideally, phase III trials should be undertaken after the foundation for the trial has been rigorously prepared, given its high cost and lengthy duration (refer to Table 3). For AD, the strength of evidence to enter phase III trials has not been very robust; as demonstrating efficacy for AD is difficult in phase II trials without adopting patient numbers (often thousands) and treatment durations (often 1.5 years) typical of phase III trials. The following is a list of potential issues of prematurely initiating the pivotal trials: Insufficient testing for clinical efficacy; Over-reliance on biomarker data; Incorrect choice of drug dose; Inappropriate reliance on post hoc subgroup analyses.
Insufficient testing for clinical efficacy
Some drugs advanced into the pivotal phase III trial for AD with far less efficacy testing than in generally accepted standard practice. Amongst these are examples of drugs approved and marketed for other diseases, and then repurposed to treat AD. With these products, safety was already established, at least at the doses for the approved indications. The evidence for effectiveness to treat AD was typically based on epidemiological observations and limited clinical studies.
Selected failed clinical trial examples. Pioglitazone, used to treat type 2 diabetes, is thought to increase energy utilization in AD patients, and may also have anti-inflammatory effects. Epidemiological evidence showed up to 2.5 times increased risk of dementia in diabetic patients [27]. Supporting clinical evidence comes from two small open-label pilot studies: 32 and 42 patients, ranging from mild cognitive impairment to moderate AD with type 2 diabetes, treated for 6 months exhibited cognitive and functional improvements [56, 57]. But a phase II study with 29 nondiabetic AD patients tested for 18 months failed to show evidence of cognitive and function improvement [58]. Despite the paucity of clinical evidence, a large phase III was initiated with almost 3,500 patients tested for up to five years to determine time to onset of mild cognitive impairment-AD development in high-risk individuals (as one of its objectives). This study was terminated early based on interim analysis that showed an inadequate treatment effect [51].
Nilvadipine is used to treat hypertension, and evidence suggests that it may increase cerebral blood flow in AD patients. An epidemiological study of close to 25,000 patients linked hypertension to risk of dementia [30]; and less AD neuropathology was observed in postmortem brains of patients on antihypertensive medication [59]. The strongest clinical evidence to support efficacy came from an open-label study of 85 patients treated over a short 6 week period, which reported improvements on cognitive tasks and executive function [60]. Based on this evidence, a phase III trial was initiated with over 500 mild-moderate AD patients for 1.5 years, which failed to demonstrate efficacy [50].
Verubecestat (BACE inhibitor) advanced into the pivotal trials without proper testing for efficacy and safety. Following two (24 to 80 patients) phase I studies [61, 62] to measure pharmacokinetics, pharmacodynamics and safety, and with no phase II trial, this drug advanced directly into two late stage trials. A phase II/III trial with close to 2,000 mild-moderate AD patients treated over 1.5 years was terminated early, with no signs of efficacy and evidence of treatment associated adverse events [45]. A phase III study of almost 1,500 prodromal patients that was initiated after the phase II/III began, was also stopped early due to signs of worsening cognition and function [46].
Over-reliance on biomarker data
Some studies relied on and accepted changes in biomarker levels (Aβ or tau) as surrogate clinical endpoints, without evidence of confirmatory changes on cognitive/functional/global performance when deciding to advance to the pivotal trial. This is despite the fact that these performance tests are the primary clinical measure in phase III trials.
Selected failed clinical trial examples. A phase I trial of AD patients treated with lanabecestat (BACE inhibitor) showed decreased CSF and plasma Aβ levels as hypothesized, but measures of clinical demonstration of efficacy was not pursued in the study [63]. The investigators then advanced into a phase II/III study of over 2,200 prodromal-mild AD patients treated over two years, and a phase III with over 1,700 mild AD patients with treatment duration of 1.5 years. Futility analyses showed that this drug would miss the trials’ cognition efficacy endpoint, and both trials were terminated [44].
A small phase II study (51 patients) with semagacestat (γ-secretase inhibitor) showed significant impacts on plasma amyloid levels as expected, but no differences on cognitive or function measures [64]. Yet two large phase III trials were initiated (over 2,600 total mild-moderate AD patients treated for 1.5 years), and were terminated early as a result of adverse events, lack of efficacy and worsening of functional ability at the higher dose [65, 66].
Incorrect choice of drug dose
In some cases, the proper drug dose(s) was not adequately established prior to commencement of phase III trials. Phase II trials usually provide robust rationale for the therapeutically effective dose range, that provides the maximum efficacy signal without adverse effects.
Selected failed clinical trial examples. A phase III study of gantenerumab (thought to clear amyloid plaques), with almost 800 prodromal AD patients treated over two years, was stopped early based on futility analysis. The sponsors reported that the selected doses were too low, and dose-dependent effects were observed such that “higher dosing with gantenerumab may be necessary to achieve clinical efficacy” [40].
Very recently, interim analysis of two parallel phase III trials (over 3,200 total early AD patients treated for 1.5 years) of aducanumab (clears amyloid plaques) reported futility. Upon subsequent analysis of the larger dataset involving three doses, effectiveness in a subset of patients receiving the high dose was reported [12]. Although this is not an example of clinical failure, it illustrates that proper dosing was not firmly established prior to phase III trials.
In a somewhat related example, the phase II/III trial with tricaprilin (increases brain energy utilization) with over 400 mild-moderate AD patients over 26 weeks failed, reportedly due to use of a new formulation which did not adequately deliver the effective dosage [67]. Adequate testing was not performed on the new formation to determine proper dosing prior to initiation of the trial.
Inappropriate reliance on post hoc subgroup analyses
With some AD drugs, reliance on post hoc subgroup analyses of failed trial data without further verification of the results led to the initiation of phase III trials that ultimately failed. It is common practice to scrutinize negative trial results to detect treatment responsive subgroups and other insights for the design of future trials. However, there is substantial risk of spurious results with such analyses [18], as subgroups have not undergone the same rigors of recruitment and randomization as the original groups, are subject to smaller sample sizes, and multiple statistical comparisons violate the assumptions implicit in statistical tests, increasing the likelihood of spurious “significant” results.
Selected failed clinical trial examples. Tarenflurbil is the R-enantiomer of the marketed non-steroidal anti-inflammatory drug flurbiprofen, with the key putative MOA of γ-secretase inhibition. A phase II trial of mild-moderate AD patients did not demonstrate efficacy, but analysis of the negative data suggested treatment benefits on functional performance in mild AD patients with the higher dose [68]. Two phase III trials were planned with mild AD patients. One study with almost 1,700 patients tested for 1.5 years failed to replicate the earlier findings [69]. The other with 800 patients was terminated early due to the negative findings of the first study.
A phase II study of bapineuzumab (inhibits amyloid plaque formation) did not demonstrate overall efficacy, but exploratory analyses showed potential efficacy in APOE ɛ4 non-carriers [70]. Subsequently, four phase III trials were initiated in mild-moderate AD patients with either APOE ɛ4 carrier or non-carrier status. Two of these trials were completed with either APOE ɛ4 carriers or non-carriers (over 2,400 patients, combined) followed for 1.5 years; efficacy was not demonstrated irrespective of APOE carrier status [71]. The other two trials were terminated early because of the reported failures of the first trials [72].
Pivotal trial design issues
We highlight phase III clinical trial design issues, where optimal drug development practices were not followed: Poor choice of primary clinical outcome measures; Inclusion of non-AD patients; Insufficient accounting for potential AD subtypes; Therapeutic interventions administered too late.
Poor choice of primary clinical outcome measures
The primary endpoint in clinical trials must be appropriate and sensitive to adequately measure treatment effectiveness in the selected patients. The most commonly used primary clinical outcome measure for cognitive performance in AD dementia patients, within the timeframe of our evaluation was ADAS-Cog. This was developed in the 1980s and has shown to measure cognitive decline in mild-moderate AD patients. However, as AD trials have targeted patients earlier in the disease progression, the sensitivity of this test has been questioned for milder affected patients [73]. An analysis of multiple studies showed that 9 of 11 ADAS-Cog subtests suffered from a ceiling effect, as these were too easy for patients with milder symptoms of AD. Alternate tests such as AD Composite Score or ADCOMS [74], comprised of selected items from other tests, is reported to be more sensitive to change and treatment effects in earlier stage patients.
Selected failed clinical trial examples. ADAS-Cog was the primary clinical outcome measure (solely or as co-primary) in 64% (32 of 50) of the failed disease-modifying phase III trials since 2004, of which 25% (8 of 32) targeted either prodromal and/or mild AD patients. Examples of such failed trials where ADAS-Cog was used in prodromal and/or mild patients include the direct amyloid impacting drugs solanezumab [39], azeliragon [49], lanabecestat [44], and tarenflurbil [69]. Supplementary Table 1 outlines the details for all phase III failures.
Inclusion of non-AD patients
As clinical diagnosis of AD is not always sufficient confirmation of the disease, biomarker measurements are useful in its verification for clinical trial inclusion. In clinical practice, there have been reports of false positives in approximately 15% of cases [75]. The misdiagnosis may involve other types of dementias such as vascular, frontotemporal and Creutzfeldt-Jakob disease, and may even involve depression and brain trauma [75, 76]. Although misdiagnosis is much less common with advancements in AD diagnosis and biomarker utilization, the possibility remains. Recently, a new form of dementia that “mimics” AD dementia has been hypothesized [77], called limbic-predominant age-related TDP-43 encephalopathy, which can be another potential source of misdiagnosis.
Subjects without AD included in clinical trials will lead to an increase in difficulty demonstrating efficacy of treatment. Misdiagnosed patients in the placebo group may not demonstrate the expected disease progression changes over time, and patterns in decline of cognitive performance, while misdiagnosed patients in the treatment group may not show the expected response to treatment.
Selected failed clinical trial examples. In 36% (18 of 50) of the phase III trial failures since 2004 of disease-modifying drugs, no biomarkers were used as part of the inclusion screening criteria to confirm AD diagnosis and exclude non-AD patients (see Supplementary Table 1). The following are examples where no biomarkers were used to both screen patients and monitor their response to treatment: nilvadipine, a calcium channel blocker thought to increase vascular blood flow, although APOE status was measured [50] (a sub-study to measure CSF Aβ and tau was discussed but no results were presented [78]); simvastatin, an anti-cholesterol medication [79]; and antibiotics doxycycline and rifampicin, with putative MOA of reduction in neuroinflammation, and also of amyloid and tau accumulation and neurotoxicity [80].
Insufficient accounting for potential AD subtypes
As evidence mounts that AD is a heterogeneous disease and may encompass different disease subtypes, this should be appropriately accounted for within the trials and perhaps subtypes should even be specifically targeted. A recent review states there may be as many as four distinct subtypes of AD [4]. One study [81] identified two subtypes of AD patients that differed significantly in the pattern of brain pathology, biomarker positivity (Aβ and tau), APOE ɛ4 carrier status, and differential scoring on cognitive subtests (memory, language, and executive function). The fact that the subtypes differ on types of cognitive test performance is of practical significance, as these test results are the primary indicator of drug effectiveness within clinical trials. AD subtypes are a potential source of variability within clinical trials, which can create variance in outcomes, lowering the signal to noise ratios.
Selected failed clinical trial examples. To our knowledge, across the failed clinical trials, none have a priori looked for differential scoring on cognitive tasks based on brain pathology patterns, biomarker positivity (Aβ and tau), and APOE ɛ4 carrier status taken together. This is not surprising, given that the recognition of subtypes has only been possible quite recently with the improvements in biomarkers and other measurement techniques.
Therapeutic interventions administered too late
There is the growing recognition that amyloid-directed interventions should occur prior to significant build-up of amyloid plaques [19]. Once the various neurodegenerative disease processes are active and progressing, prevention of further Aβ formation or removal of existing plaques, and other degenerative processes may not be as effective. We do not know at this time how early is early enough, especially given the understanding that initial changes in the brain may occur many years before the first clinical manifestations of the disease [82, 83]. We know that AD prevalence rises sharply after age 65 years, and 97% of cases are after this age [1]. Although there is no direct correlation between an individual patient’s age and stage of the disease, and that cognitive performance scores are a better indicator of stage of disease, generally speaking early-stage patients are a younger population. Thus, as a general rule, younger patients should be recruited into trials, all other factors being equal. Furthermore, focusing on milder impacted populations (e.g., prodromal) based on cognitive measures may yield better results. However, it should be kept in mind that the inclusion of earlier stage AD patients may lead to more misdiagnosis, as the symptoms are more subtle.
Selected failed clinical trial examples. In many clinical trials (refer to Supplementary Table 1), the lower end of the patient age range is typically 50–55 years, and the upper end is 85–90 years or no upper limit. This is the norm even when prodromal or mild AD is targeted, as is the case with the failed amyloid targeting drugs elenbecestat [41], lanabecesat [44], crenezumab [47], solanezumab [39], and gantenerumab [40]. Some sponsors have explicitly stated in phase III post-failure communications that intervening too late in the disease process may be a reason for the failure, i.e., lanabecestat [44], crenezumab [84], and bapineuzumab [85]; all of which tested AD patients ranging from prodromal to moderate.
RECOMMENDATIONS FOR FUTURE AD THERAPEUTICS DEVELOPMENT
With the benefit of “perfect” hindsight (we know much more now than when these trials were initiated), our evaluation showed that rational drug development principles were not always followed for AD. Phase III studies were initiated despite insufficient preliminary evidence, and the phase III trial designs were less than optimal. Adherence to more stringent criteria for progression into the pivotal trials follows the maxim of failing early, allowing the shift of resources to other promising compounds. For pivotal trials, better choice of clinical outcome measures, utilization of multiple biomarkers for patient screening and identification to better account for non-AD patients and AD subtypes, and targeting patients earlier in the disease progression may lead to greater trial success. With AD, sponsors may be tolerating high R&D risk because of the high unmet medical need (disease severity and dearth of effective treatments), notable difficulty in establishing efficacy, and the huge financial reward for an effective treatment.
We cannot and do not state that the factors outlined are directly responsible for the clinical trial failures; these are highlighted as potential contributing factors for the negative trial results. The compound failures may have resulted because they are simply ineffective, the MOA is not relevant, beneficial effects may be neutralized due to other pathways being impacted, etc. However, is it possible that some of the failed compounds could have shown efficacy with adherence to more robust drug development principles? We cannot answer this with our current evaluation, but it does pose the question of whether any of the failed compounds should be re-examined with better designed clinical trials.
Even the most optimized drug development programs are limited by our incomplete understanding of AD’s underlying pathophysiology with its multiple biologic pathways and therapeutic targets, and also the state of the science guiding the potential therapeutic interventions. Given the current gaps in our knowledge, we offer selected suggestions to guide future AD therapeutics development. Other researchers have also proposed many different future directions [22, 86].
Beyond the amyloid hypothesis
According to the amyloid cascade hypothesis, Aβ is the primary causative agent in AD pathology, and the neurogenerative effects and clinical symptoms are downstream manifestations [87]. This view has guided much of the therapeutic efforts for almost three decades, but some have begun to question the nature of the relationship between Aβ and AD [88, 89]. There are many reported dissociations between levels of brain Aβ and AD clinical diagnosis [25, 90]. It is not uncommon for brains of elderly cognitively normal individuals to have the load and distribution of senile plaques that satisfy the criteria for AD. Conversely, some AD patients clinically diagnosed by the current available tools have few brain amyloid deposits.
This disassociation was also observed in our evaluation of the 23 failed AD compounds (∼ one quarter of the total) that targeted Aβ at multiple points of the physiologic pathway. There were multiple instances of changes in Aβ levels confirming target engagement, without corresponding improvements in cognitive function. For example, in failed phase II/III or III trials, bapineuzumab (inhibits amyloid plaque formation) reduced PET Aβ [71, 85]; verubecestat (BACE inhibitor) lowered PET Aβ and CSF levels of Aβ40 and Aβ42 [45, 46]; and lanabecestat (BACE inhibitor) reduced CSF levels of Aβ40 and Aβ42 [44].
The efficacy of oligomannate [16], should it hold up to further medical scrutiny, may provide a potential non-amyloid target for therapeutics of intestinal microbiomes and anti-inflammatory actions (although earlier evidence suggested Aβ is impacted as well [91]). The recent reported efficacy of aducanumab (monoclonal antibody engineered to clear amyloid plaques) with demonstrated target engagement (decrease in Aβ deposition) seems to support the amyloid hypothesis [12]. However, there were some notable issues with the evidence: efficacy was demonstrated in only one of the two phase III studies and was not replicated in a further phase III; a subgroup was selected to illustrate proof of efficacy without further verification; lack of correlation between biomarker and clinical data; data were missing from the early termination of the studies; and potential confounding by selective patient dropouts due to amyloid-related imaging abnormalities [92].
At this time, the bulk of evidence still supports a relationship between Aβ and AD, but the relationship is probably more complex and nuanced than originally postulated. There is an emerging view that Aβ may be necessary, but is not sufficient in itself to cause AD [89, 93]. Aβ is likely a key initiator of a complex cascade; that it acts primarily as a trigger of other downstream processes, but other physiological conditions must be present for AD to develop. Examples of alternate evidence suggests that the trigger of AD is closely linked to impairments of Aβ protein precursor metabolism and accumulation of its fragments, more so than Aβ, and that tau may play a greater role in AD than Aβ [25, 90].
The amyloid hypothesis is one of many (albeit the most prominent one); and other hypotheses and therapeutic approaches should continue to be actively pursued. As one author summarized, “the continued push toward a safe and efficacious amyloid therapeutic takes nothing away from the need for alternative agents that target other early features of this complex and devastating syndrome” and “it is not a question of one hypothesis against another” [5].
Address AD heterogeneity
As discussed above, recent advances highlight AD to be a heterogeneous disorder, with the distinction between early versus late onset variants, and the potential for multiple subtypes of late onset AD. With the late onset variant, two distinct subtypes were distinguished by the pattern of brain pathology, biomarker positivity, APOE carrier status, and cognitive test results [81]; three subtypes were differentiated on the bases of metabolic profiling [94]; as many as four types based on patterns of brain atrophy [95, 96]; and a review article made a case for four subtypes [4].
AD heterogeneity, in ways that need further characterization, may lead to different patterns of symptomatology and disease progression, potentially resulting in differential treatment response in symptoms, effect sizes and time-course over the duration of treatment. It may not be realistic for a single drug to treat such heterogeneity in equal measure. Future clinical trials should, at least, track subtype distributions across the trial arms, specify a priori intent of subset analyses, or be designed to specifically target certain subtypes (beyond just APOE status); and at best, adopt a personalized medicine approach to AD therapeutics development.
Multiple biologic targets
We are becoming increasingly aware of the complexity and multifaceted nature of this disease. For example, AD neurodegeneration is linked to dysregulation of cholesterol homeostasis, changes in energy metabolism and mitochondrial dysfunction, activation of neuroinflammatory pathways, and the role of neurotrophic factors within the brain [6–8, 34]. There are also interactions between the brain and other physiological systems, demonstrated by the established links with pathological conditions such as inflammatory disorders, hypercholesterolemia, hypertension, and diabetes.
Given such complexity and the involvement of so many other physiological systems, a multi-target intervention approach may be more effective than those working via a single target [97, 98]. We have already witnessed the failure of almost a hundred predominantly single target compounds over the past two decades. The classic ‘one-drug/one-target/one-disease’ approach seems insufficient [98]. Simultaneously aiming at different targets within the pathophysiology of the disease may lead to synergistic therapeutic effects and thus better overall efficacy.
Support for this view is from a multifactorial modelling approach to AD pathology and therapeutic intervention [99]. The model showed that AD may not be caused by a unique dominant biological factor (such as Aβ) but by the complex interplay amongst multiple relevant direct interactions; and that the combinatorial approach to treatment should outperform singular therapies. This model still requires further validation at the individual patient level; nevertheless, it does present an analytic framework for the dynamic multifactorial brain organization and the potential effectiveness of multi-mode interventions. Of course, this approach has additional potential challenges such as multiple-drug interactions, compounding of adverse effects, and increased costs of clinical trials due to multiple dose combinations of the two or more drugs.
Protective factors
As discussed above, neurodegenerative changes begin in the brain many years, perhaps even decades prior to clinical manifestations of AD [82, 83], and some researchers believe that intervening too late in the disease process may be a reason for phase III failures with lanabecestat [44], crenezumab [84], and bapineuzumab [85].
It would be a very difficult endeavor to develop prophylactic therapeutic interventions in individuals with no clinical symptoms of the disease, when any protective benefits are not realized for several decades. However, it may be easier to place more emphasis on controlling known risk factors for AD, especially prior to manifestations of clinical symptoms. Acquired factors such as cerebrovascular diseases, diabetes, hypertension, obesity, and dyslipidemia are known to increase development of AD [6, 100]. As these are predominantly lifestyle disorders linked to diet, lack of exercise, etc., controlling such behaviors should have a protective effect in AD development [100]. Given the difficulty in changing these types of behaviors, finding ways to make lifestyle modifications more broadly adopted by large populations in a sustained way should be a public health priority.
Systems approach using artificial intelligence and quantitative systems pharmacology
There has been a notable lack of success in AD therapeutic development with the current, generally reductionistic approach to drug development. A systems approach that considers the organism holistically may yield better results. This would incorporate the full complexity and involvement of multiple physiological pathways and brain-body interactions, to overcome the overreliance on single pathway and single target approaches. A quantitative systems pharmacology (QSP) model combined with artificial intelligence (AI) would be informed by data generated from observation and research studies, as well as clinical trials. This approach could identify gaps in our knowledge of the disease, generate new biological or pharmacological hypotheses, and aid in the design of in vitro and in vivo experiments to investigate and validate the model insights.
Such modelling approaches have yielded better understanding of the interplay of different physiological systems with pathological conditions and even in silico clinical trials [101–103]. Recently, scientists have begun to apply modelling approaches to AD with encouraging results. For example, AI models have shown great accuracy at diagnosing AD patients [104]. And as mentioned already, a multifactorial model of AD pathology and therapeutic intervention [99] provided evidence that AD may not be caused by a unique dominant biological factor but by the interplay of multiple relevant direct interactions; and the combinatorial approach to treatment should be superior to singular therapies.
We propose an approach that synthesizes large datasets of the latest scientific and clinical information, and AD clinical databases such as Alzhei-mer’s Disease Neuroimaging Initiative and Rush Alzheimer’s Disease Center. These are extensive databases of patients with AD and without dementia tracked with various metrics over extended periods of time. It contains both structured measurements (cognitive assessments, biomarker, genetic information) and a variety of imaging information (magnetic resonance imaging, PET, etc.). The latest explainable AI models may be used to extract meaningful imaging biomarker information to supplement the longitudinal biomarker information and progression of cognitive decline. With the QSP-type approach, the current information on pathways implicated in the pathology of AD may be used to explain the heterogeneity in disease progression. The primary challenge will be explainability of the many failed clinical trials, and the lack of ability to reproduce such failure would highlight the gaps in the current understanding of the disease mechanisms. Such a platform may likely aid in finding new targets or combination of targets for specific cohorts of AD patients to maximize the chances of demonstrating clinical efficacy. Importantly, this overall approach should lead to the generation of testable hypothesis that can be taken into animal models and human trials.
CONCLUSIONS
AD is perhaps the most serious unmet medical need today. Notwithstanding two very recent developments (aducanumab and oligomannate), which still require real-world validation of efficacy, 2003 was the last year of a novel drug approved for AD. In order to better understand the reasons for such unprecedented lack of success, we performed a review of clinical trial failures for AD from 2004 to the present. The objective was to derive key insights from these myriad failures to guide future drug development in AD.
Of the ∼2,700 clinical trials for AD within ClinicalTrials.gov, ∼550 interventional phase II and III trials examined cognitive performance, in which 98 unique phase II and III compounds failed. Given the timing of this analysis, we present the most recent AD drug development success rate of 2.0% (since the last previous novel compound success).
Our evaluation found that the principles of rational drug development were not always followed. Phase III studies were initiated with: Insufficient confirmatory evidence of clinical efficacy; Over-reliance on biomarkers as a surrogate for clinical efficacy; Insufficient testing to determine effective drug dose(s); Lack of confirmation of post hoc subgroup analysis before committing to the pivotal trial. Phase III trial designs were less than optimal as: Primary clinical outcome measures chosen for cognitive performance was not ideal for the targeted patient cohorts; Biomarkers were not always included as part of the trial design to confirm accurate diagnosis and exclude non-AD patients; Insufficient accounting for the AD subtypes; Therapeutic interventions were occurring too late in the disease progression continuum.
These insights from the evaluation of clinical trial failures prompted the question of whether some of the failed compounds might have shown efficacy if more robust drug development principles had been followed; and if some of these failed compounds warrant re-examination with better designed clinical trials. At this time, we cannot quantify the relative contributions of the faulty drug development process versus the lack of understanding of the underlying pathophysiology of AD (biological pathways, targets, robustness of therapeutic interventions, etc.). While the research progresses to fill in the scientific gaps, there are approaches to therapeutic development that we can adopt now: The amyloid hypothesis which has predominantly guided R&D for almost three decades is proving to be less robust, and so other hypotheses and therapeutic approaches should continue to be actively pursued to increase the overall chance of finding effective treatments. It may not be realistic for a drug to effectively treat the whole spectrum of AD subtypes, given that there may be up to four subtypes of late onset AD, in addition to the early onset variant. Thus, future trials should target specific subtypes which are most likely to respond to the specific therapeutic intervention, and generally adopt a more personalized medicine approach to drug development. Given the noted lack of success with the predominantly single target approach, compounds aimed at multiple targets simultaneously with synergistic effects should be prioritized. More effort should be given to reduce the risk factors for AD by reducing certain pathologies (e.g., cardiovascular, metabolic, etc.) that contribute to AD. These are partially lifestyle disorders, and thus preventable to a large degree with diet and exercise. Finally, as we progressively appreciate the complexity and multifaceted nature of this disease with the interplay between the brain and many other physiological systems, a systems approach using AI and QSP modelling, that considers the organism holistically may yield better results. We should move away from the generally reductionistic approach that we have so far relied on.
Despite the many advances in recent years, we currently do not have a cure or validated treatment that effectively stops AD progression, thus there is much work to be done. Adherence to more stringent drug development principles and adopting very different approaches and thinking of AD treatments may ultimately lead to the successful development of more effective treatments for this debilitating disease. Learnings from the clinical trial failures and the efforts of countless patients and caregivers who participated in these trials is a strong base from which to build as we progress forwards. There is no doubt that the ingenuity of medical science will prevail; it is a matter of when, not if.
Footnotes
ACKNOWLEDGMENTS
Dr. Jeffrey Cummings (Cleveland Clinic Lou Ruvo Center for Brain Health, NV, USA) provided invaluable comments and feedback on the paper.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.