
Abstract
Acute delirium and other neuropsychiatric symptoms have frequently been reported in COVID-19 patients and are variably referred to as acute encephalopathy, COVID-19 encephalopathy, SARS-CoV-2 encephalitis, or steroid-responsive encephalitis. COVID-19 specific biomarkers of cognitive impairment are currently lacking, but there is some evidence that SARS-CoV-2 could preferentially and directly target the frontal lobes, as suggested by behavioral and dysexecutive symptoms, fronto-temporal hypoperfusion on MRI, EEG slowing in frontal regions, and frontal hypometabolism on 18F-FDG-PET imaging. We suggest that an inflammatory parainfectious process targeting preferentially the frontal lobes (and/or frontal networks) could be the underlying cause of these shared clinical, neurophysiological, and imaging findings in COVID-19 patients. We explore the biological mechanisms and the clinical biomarkers that might underlie such disruption of frontal circuits and highlight the need of standardized diagnostic procedures to be applied when investigating patients with these clinical findings. We also suggest the use of a unique label, to increase comparability across studies.
INTRODUCTION
Acute delirium and other neuropsychiatric symptoms have frequently been reported in COVID-19 patients, either along the disease course or as first symptoms [1]. This clinical presentation has variably been referred to as acute encephalopathy, COVID-19 encephalopathy, SARS-CoV-2 encephalitis, or ster-oid-responsive encephalitis [2–4]. The different terminology used reflects the relative lack of identified and consistent pathognomonic findings, such as neuropathological, biochemical, and imaging bio-markers. However, by inconsistently conceptualizing the pathogenesis of similar presentations using proteiform labels, we risk missing the big picture of a common shared neurobiological entity. It is important to search for common and shared clinical and imaging data that could improve the management of patients suffering for these complications of COVID-19 infection. Indeed, given the high percentages of COVID-19 patients with acute delirium, and poor shared COVID-19 biomarkers and diagnostic protocols, we are in need of a unifying model, to increase comparability across studies. This hypothesis proposes a re-thinking of these discrete subtypes, in light of common biological findings, which point toward a selective vulnerability of the frontal lobes to SARS-CoV-2 infection.
CLINICAL EVIDENCE
As reported both in case series and large cohort studies [2–4], delirium and frontal-type behavioral changes have often been described in COVID-19 patients; notably these symptoms can be associated with motor signs, such as corticospinal signs, myoclonus, rigidity, and akinetic mutism (Fig. 1, Table 1). Furthermore, cognitive impairment with prominent decline in executive functions has been reported in COVID-19 patients, either in the acute phase, or at discharge, and correlates with COVID-19 disease severity and use of mechanical ventilation [2, 6]. Presence of inflammatory markers in serum and/or cerebrospinal fluid (CSF), such as increased interleukin 6 (IL-6), IL-8, or oligoclonal bands, have been reported by several groups in association to this clinical phenotype [2–4, 7]. Frontal abnormalities on electroencephalography (EEG), including slow-waves and slowing-down of the background rhythm or epileptiform discharges, are present in one-third of COVID-19 patients and have been proposed as an EEG biomarker for COVID-19 encephalopathy [8]. In these patients, brain imaging using magnetic resonance imaging (MRI) may be unremarkable or show signs of frontotemporal hypoperfusion with arterial spin labeling [2, 3]. Also, frontal hypometabolism on positron emission tomography with F-18 fluorodeoxyglucose (FDG-PET) has been found in COVID-19 patients with acute frontal-type behavioral changes [9, 10], as well as in patients without acute delirium, but with symptoms such as anosmia or ageusia [11, 12]. In several cases, counteracting inflammation with steroids, IVIG, or Tocilizumab has resulted in normalizations of symptoms and EEG findings in patients with acute delirium and biomarkers of frontal dysfunction [4, 23].
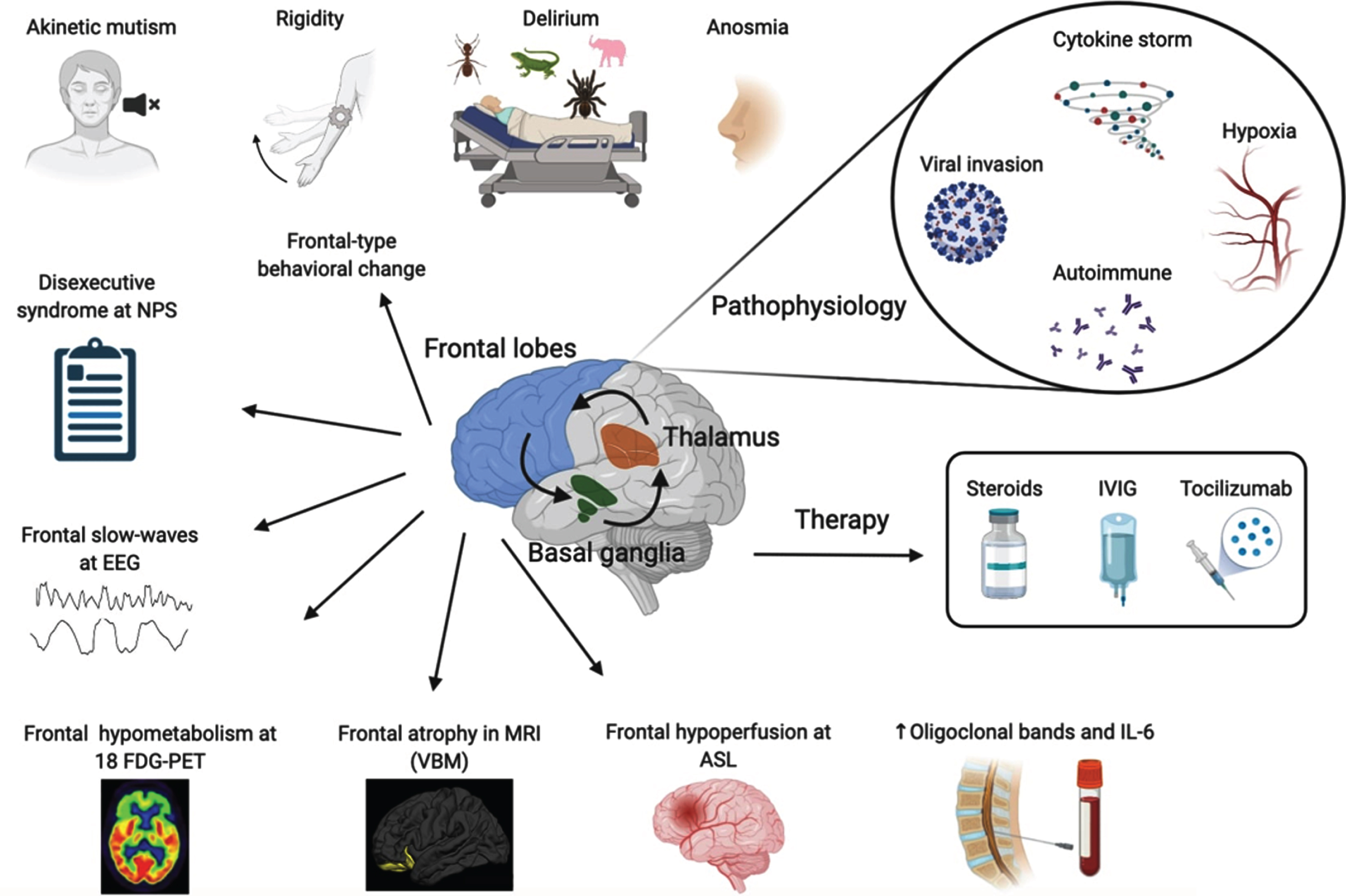
Overview of the pathophysiological mechanisms, symptoms, biomarkers of frontal lobe encephalopathy and therapy in COVID-19 patients. NPS, neuropsychological tests; EEG, electroencephalogram; 18 FDG-PET, positron emission tomography with F-18 fluorodeoxyglucose; MRI, magnetic resonance imaging with voxel-based morphometry; ASL, arterial spin labeling; IL-6, interleukin 6; IVIG, intravenous immunoglobulins. Made in ©BioRender - biorender.com.
Overview of the studies
N, number of patients; MV, mechanical ventilation; WM, white matter; EEG, electroencephalogram; CRP, C-reactive protein; IVIG, intravenous immunoglobulin.
Even if heterogeneity of clinical presentations has been reported, there are several features that are striking in their commonality across these cases [14]. We hypothesize that the relative association of these findings could point toward a common pathophysiological mechanism, and these cases could possibly belong to the same nosological entity. The combination of the abovementioned data suggests a vulnerability of the frontal lobe or, at least, of the frontal hubs of cortical-subcortical networks to SARS-CoV-2 infection. This would be in keeping with a disruption of fronto-striato-thalamo-cortical circuits seen generally in akinetic mutism [15]. Indeed, the main hubs involved in akinetic mutism are the prefrontal cortex, the thalamus, and the striatum, where ACE-2 receptors, the major entry route for SARS-CoV-2, are preferentially expressed [15–17]. Neuropathological data suggest that these regions might be affected by SARS-CoV-2, albeit a lower concentration of cytotoxic T lymphocytes can be detected in the frontal lobes and basal ganglia if compared to the brainstem, one of the key targets of SARS-CoV-2 [18]. An acute inflammatory encephalopathy presenting as malignant catatonia in a COVID-19 patient, with involvement of the striatum, has been reported [19], further suggesting a vulnerability of this circuitry to COVID-19 infection.
PATHOPHYSIOLOGICAL THEORIES
The high prevalence of COVID-19 neurological symptoms highlights the potential neurotropism and neurovirulence of SARS-CoV-2, as seen with other neurotropic human coronaviruses [20]. However, there is still debate regarding their putative underlying mechanisms and different theories have been put forward.
The first hypothesis is that these symptoms are driven by cytokine-mediated neurotoxicity. It has been suggested that COVID-19 patients could develop a form of cytokine-mediated neuroinflammatory encephalopathy, the immune effector cell–associated neurotoxicity syndrome (ICANS), due to the massive cytokine storm induced by COVID-19 systemic infection [13]. ICANS is a neuropsychiatric syndrome that may include frontal-type changes, such as confusion, short-term memory impairment, expressive deficits, and behavior disturbances including impulsivity, emotional lability, abulia, or akinetic mutism as clinical phenotype [13, 22].
A second theory is that this syndrome is driven by an autoimmune process. The presence of oligoclonal bands and the strong response to immunomodulatory treatments might support its dysimmune origin [4, 23]. The few neuropathological studies in COVID-19 patients available at the present time support this theory, as they show signs of neuroinflammation [18]. Indeed, the pattern of activated microglia seems reminiscent of autoimmune encephalitis rather than direct central nervous system (CNS) damage caused by the virus [18]. An inflammatory process could be in keeping with increased levels of glial markers, including fibrillary acidic protein (GFAP), sTREM-2, and YKL-40, found in COVID-19 patients in plasma and in the brain [7, 24]. COVID-19 encephalitis presenting as Acute Disseminated Encephalomyelitis (ADEM) or limbic encephalitis have also been reported, suggesting a possible, though rarer, autoimmune mechanism [25–27]. Vascular involvement as intracerebral endotheliitis has also been extensively described, resembling vasculitic changes caused by SARS-CoV-2 in lungs, skin, and across the body [25, 29].
A third proposition postulates a primary invasion of the CNS. Even though positivity for SARS-CoV-2 on PCR in the CSF is relatively rare, SARS-CoV-2 RNA can be detected in the frontal lobes in these patients [18, 30], probably through invasion of the olfactory bulb, with spreading to the anterior cingulate and basal forebrain. The involvement of these structures could potentially explain the presence of frontal symptoms, and particularly an akinetic mutism presentation [15].
Another proposed pathogenic mechanism is hypo-xia. Acute respiratory distress syndrome (ARDS) is able to induce hypoxia, that in turn would produce a hypoxic encephalopathy, especially in older patients with high vulnerability to oxidative stress. Frontal lobe abnormalities secondary to hypoxia have been described in patients who received mechanical ventilation [31]. However, not all the patients with SARS-CoV-2 patients who have developed these encephalopathic symptoms were admitted to intensive care units (ICU) and received invasive mechanical ventilation, and therefore could not be the only causative explanation. Moreover, the neuropathological findings in COVID-19 patients show a widespread neuroinflammatory pattern, while a pure hypoxic ischemic injury is the hallmark of patients with delirium admitted to ICU and treated with mechanical ventilation for other causes rather than COVID-19 infection [31].
These models are not mutually exclusive, and an invasion, even minimal, of the virus in the CNS, might induce mild reactive neuropathological changes that would make, selectively, neurons more vulnerable to a systemic cytokine storm, causing increased oxygen free radicals, and eventually leading to neuronal dysfunction and death.
CONCLUSION
We suggest that an inflammatory parainfectious process targeting preferentially the frontal lobes (and/or frontal networks) could be the underlying cause of these shared clinical, neurophysiological, and imaging findings in COVID-19 patients. Indeed, this constellation of findings seems specific for SARS-CoV-2 as it has not been described in other viral infections, such as influenza A (H1N1), where encephalopathy is a rare complication, mostly affecting children, and does not show similar symptoms and imaging features related to a frontal lobe dysfunction [32]. Albeit MERS and SARS-CoV-1 could present with neuropsychiatric symptoms, the limited data available on neuroimaging and biomarkers do not support similar syndromic changes [20].
A larger collection and description of these cases through multicentric studies is needed to understand the clinical and biological biomarkers of this entity as we cannot exclude that only a subset of the patients in each study share the same pathophysiological underpinnings. Standardized diagnostic procedures should be applied when investigating patients with these clinical findings. Neurologists should be part of the multidisciplinary team assessing COVID-19 patients, for early detection and reliable clinical evaluation of COVID-19-related neurological symptoms and signs [33]. When possible, neuropathological investigations should be performed, to allow a better characterization of the macroscopic and microscopic morphological alterations and help in the identification of a common pathophysiological disease substrate. International platforms such as the European Academy of Neurology COVID-19 registry (ENERGY), a large multicentric tool for surveillance of neurological complications in COVID-19 patients, might provide a useful aid for clinical data sharing and large scale studies, which will be essential for understanding this new entity [34]. Moreover, we highly advise to follow established recommendations on how to report neurological manifestations of SARS-CoV-2 infection to help reducing reporting bias [35]. Thus, the use of a unique label is needed to increase comparability across studies, and given that direct CNS damage is not ubiquitous, and its etiology multifactorial, the term COVID-19 associated frontal lobe encephalopathy might be preferred to define SARS-CoV-2 indirect manifestations targeting the frontal lobe.
Footnotes
ACKNOWLEDGMENTS
We would like to thank the Dementia Panel of the EAN (European Academy of Neurology), and their COVID-19 task force for inspiring this work. This research received no specific funding from any agency in the public, commercial, or not-for-profit sectors.
All authors are members of the EAN Dementia and Cognitive Disorders Scientific Panel.