
Abstract
Alzheimer’s disease (AD) is a multifactorial syndrome with a plethora of progressive, degenerative changes in the brain parenchyma, but also in the cerebrovascular and hemostatic system. A therapeutic approach for AD is reviewed, which is focused on the role of amyloid–β protein (Aβ) and fibrin in triggering intra-brain vascular dysfunction and connected, cognitive decline. It is proposed that direct oral anticoagulants (DOACs) counteract Aβ-induced pathological alterations in cerebral blood vessels early in AD, a condition, known as cerebral amyloid angiopathy (CAA). By inhibiting thrombin for fibrin formation, anticoagulants can prevent accumulations of proinflammatory thrombin and fibrin, and deposition of degradation-resistant, Aβ-containing fibrin clots. These fibrin–Aβ clots are found in brain parenchyma between neuron cells, and in and around cerebral blood vessels in areas of CAA, leading to decreased cerebral blood flow. Consequently, anticoagulant treatment could reduce hypoperfusion and restricted supply of brain tissue with oxygen and nutrients. Concomitantly, hypoperfusion-enhanced neurodegenerative processes, such as progressive Aβ accumulation via synthesis and reduced perivascular clearance, neuroinflammation, and synapse and neuron cell loss, could be mitigated. Given full cerebral perfusion and reduced Aβ- and fibrin-accumulating and inflammatory milieu, anticoagulants could be able to decrease vascular-driven progression in neurodegenerative and cognitive changes, present in AD, when treated early, therapeutically, or prophylactically.
Keywords
INTRODUCTION
Early clinical trials with small groups of patients with senile dementia symptoms indicated a positive effect of anticoagulant treatment on disease development [1 –3]. Results of basic research in the last six years on a contribution of cerebrovascular dysfunction to Alzheimer’s disease (AD) have strengthened this idea. As reviewed in this article, I discuss how toxic proteins of amyloid–β (Aβ), thrombin and fibrin are key drivers in triggering vascular abnormalities and derived neurodegenerative changes, present in AD, which can be treated by anticoagulants.
ANTICOAGULANT EFFECT ON BLOOD CLOTTING AND THROMBOSIS
In the case of injuries to the blood vessels, excessive blood loss in the organism is prevented by the multi-stage process of hemostasis [4]. In the phase of blood clotting (coagulation), which leads to the closure of the wound and healing, a soluble protein from the blood, the fibrinogen, is converted into insoluble fibrin. Subsequently, a fibrin fiber network with integrated erythrocytes and platelets, a fibrin clot (thrombus), is formed. The synthesis of the responsible key enzyme, the serine protease thrombin, is induced cascade-like by a multitude of tissue and coagulation factors (e.g., factor Xa). Some of these factors are activated in a vitamin K-dependent process. Endogenous inhibitors counteract the excessive formation of fibrin clots. For example, the tissue plasminogen activator t-PA induces the conversion of plasminogen to the proteolytic enzyme plasmin. Plasmin degrades fibrin in the fibrinolysis process and thus, dissolves fibrin clots [4].
Hereditary changes in distinct coagulation factors or inhibitors, large wound areas after surgeries and injuries, and decelerating blood flow (e.g., through atrial fibrillation, changes in blood vessels due to atherosclerosis, surgeries, limited movement) can cause increased blood clotting with the formation of thrombi. These thrombi can close blood vessels in the process of thrombosis, or, after detachment, shift through the vascular system and can cause pulmonary embolism or brain infarction [4].
Anticoagulants are drugs that are used prophylactically or therapeutically to inhibit blood clotting and therefore, avoid the formation of thrombosis or embolisms. Anticoagulants can inhibit blood clotting in different ways [5]: indirectly, such as in the case of vitamin K antagonists (VKAs, e.g., warfarin, phenprocoumon, acenocoumarol, with oral effect), in heparins (e.g., enoxaparin), heparinoid dana-paroid sodium and fondaparinux (with parenteral effect); directly, such as in the case of thrombin inhibitors (e.g., dabigatran etexilate, ximelagatran, with oral effect; hirudin, bivalirudin, argatroban, with parenteral effect) and in blood clotting factor inhibitors (e.g., factor Xa inhibitors, such as apixaban, rivaroxaban, edoxaban, betrixaban with oral effect; otamixaban, with parenteral effect). Direct thrombin inhibitors and factor Xa inhibitors are referred to as direct oral anticoagulants (DOACs).
Short-term anticoagulation is indicated for acute treatment of venous thrombosis and thrombosis prophylaxis in risk situations, e.g., after surgeries, and in persons with limited mobility. Long-term to permanent anticoagulation is indicated, e.g., for recidive prophylaxis after venous thrombosis, for the prophylaxis of cardiac thrombosis and embolisms in patients with cardiac arrhythmias, such as atrial fibrillation, and in patients with mechanical heart valve replacement [5]. In Germany alone, anticoagulants in form of VKAs and DOACs are prescribed in approximately 1.0 and 2.0 million patients, respectively [6]. The patients are predominantly over 70 years old, due to the indication areas of anticoagulants [6]. However, a side effect of all anticoagulants is that their anticoagulant effect is associated with an increased risk of bleeding [5]. Conversely, anticoagulant therapy reduces the risk of a fatal heart attack or stroke in vulnerable individuals. A medication based on anticoagulants for treatment of the most common neurodegenerative brain amyloidoses, such as AD and Parkinson’s disease, is not yet in use.
ALZHEIMER’S DISEASE: PATHOPHYSIOLOGICAL HALLMARKS AND CURRENT MEDICATION FOR THERAPY
The drugs, available so far, for the treatment of AD, which includes cholinesterase inhibitors (e.g., donepezil, galantamine, rivastigmin) and glutamate antagonists (e.g., memantine), are only able to delay the onset of dementia symptoms for a certain period of time [7]. Therefore, the search for novel, more effective drugs is an urgent task of pharmaceutical research [8].
Recent research shows that accumulations of misfolded, toxic proteins of amyloid–β (Aβ) in brain tissue are crucial for triggering the AD pathogenesis [8 –18]. Aβ is enzymatically released by secretases from the amyloid–β protein precursor (AβPP). AβPP is anchored in the cell membrane of neurons and delivers fission products of different lengths (secreted AβPP) that are important for the function of synapses [19]. In contrast, toxic Aβ accumulates in form of soluble dimers and oligomers, as well as in form of insoluble, deposited fibrils (Aβ plaques). Aβ plaques and oligomers are localized between neurons in certain parenchymal tissue areas of the brain (especially in the neocortex and hippocampus: Aβ42 and Aβ43) and around and in cerebral blood vessels (in particular Aβ40). Soluble and insoluble forms of Aβ are in a complex balance. Aβ is actively transported into the blood system via the vascular interface of the brain, known as blood-brain barrier (BBB) [20]. A causal role of these Aβ accumulations in early AD pathogenesis suggests that 1) soluble Aβ aggregates led to hyperactivation and damage to neurons and synapses [18, 21], 2) all major gene modifications, associated with an increased AD risk, are related to the formation of Aβ plaques, and 3) the early treatment of AD patients with the anti–Aβ antibody aducanumab has the potential to improve cognitive performance at reduced Aβ deposition [22]. In addition, intraneural deposits and spread of tau-protein fibrils, defects in BBB, neural inflammatory processes with formation of reactive oxygen species (ROS), as well as the loss of synapses and neurons are characteristic of the progression of AD pathogenesis. Therefore, these processes are also in the focus of intensive therapeutic research [8 , 23–26]. In particular, inflammatory processes, emanating from activated microglia cells and their release of interleukins and ASC (apoptosis-associated speck-like protein containing a CARD) protein complexes in microgliosis, promote the formation and spread of cerebral Aβ deposits [27, 28]. On the other hand, activated microglia cells in the early stages of the disease led to the breakdown of cerebral Aβ deposits. Therefore, mutations in the activating gene TREM2 of microglia cells increased the formation of Aβ plaques and thus, the AD risk [29]. It is estimated that cerebral Aβ accumulates in people, who develop AD, 10 to 20 years before symptoms of the disease occur [30]. Likewise, an indicator protein for the death of brain neurons (neurofilament light chain protein, Nfl) increased in the blood of people with familiar AD, 16 years before the onset of symptoms [31].
However, less in the focus of therapeutic research so far, but also typical of the early development of AD, are Aβ-induced pathological changes in the cerebral blood vessels, a condition, known as cerebral amyloid angiopathy (CAA; Fig. 1). In the CAA, aggregates of Aβ accumulate and deposit around and into the walls of cerebral arteries and capillaries and lead to intra-brain vascular dysfunction (see surveys by [26 , 33]). Particularly, neocortical and hippocampal areas of the brain are disturbed in their blood circulation and are no longer supplied sufficiently with blood and its constituents, such as oxygen, glucose, ions, amino acids, hormones, proteins, and cellular blood components [14 , 34–36]. In addition, removal of Aβ from the interstitial brain fluid into the blood was shown to be impaired by the harmed cerebral blood vessels [37, 38]. The resulting deteriorated “blood flow and degradation clearance” of Aβ in the blood stream (perivascular Aβ clearance) promotes the accumulation of Aβ in the brain tissue and therefore, the progression of AD pathogenesis [37, 39]. Studies in AD patients and transgenic mouse lines with gene modifications in the formation of Aβ plaques for human AD risk (AD mouse models [40, 41]) showed that deposits of Aβ in the cerebral blood vessels are responsible for causing CAA disease [16 , 42–45]. Decreased cerebral blood flow (CBF) and therefore, disruption of blood circulation (hypoperfusion) of certain areas of the brain, leading to undersupply of the tissue, especially with oxygen (hypoxia) and glucose, are the consequences [14 , 47] (Fig. 1). This is followed by enhanced Aβ production via hypoxia-induced β-secretase1 (BACE1) expression [48], inflammatory and neurodegenerative changes in the brain, and cognitive decline [49, 50]. Accordingly, detailed studies in living brain tissue from human biopsies and rodent models showed that Aβ deposits, especially around cortical blood capillary vessels, cause pericyte cells on the outer wall of the vessel to contract, via ROS-induced endothelin–1 release [47]. This reduces the diameter of the capillary vessels (vasoconstriction). The CBF decreases and a chronic hypoperfusion with hypoxia is triggered [47]. In clinical studies, CAA was detected in approximately 90% of patients with AD [51]. Similarly, early in AD, cortical CBF decreases by about 25% in patients [35 , 52], as well as in AD mouse models [46, 53]. Hypoperfusion is considered as an important pathophysiological process that promotes AD [32], together with other follow-up effects of CAA (Fig. 1). These include cerebral microvascular infarctions and microhemorrhages (microbleeds), which violate BBB and promote inflammatory and degenerative changes in brain tissue [54]. Accordingly, physical activity, which causes a better blood flow to the brain, led to a slowdown in neurodegenerative processes in individuals with genetic AD predisposition [55] and in AD mouse model [56].
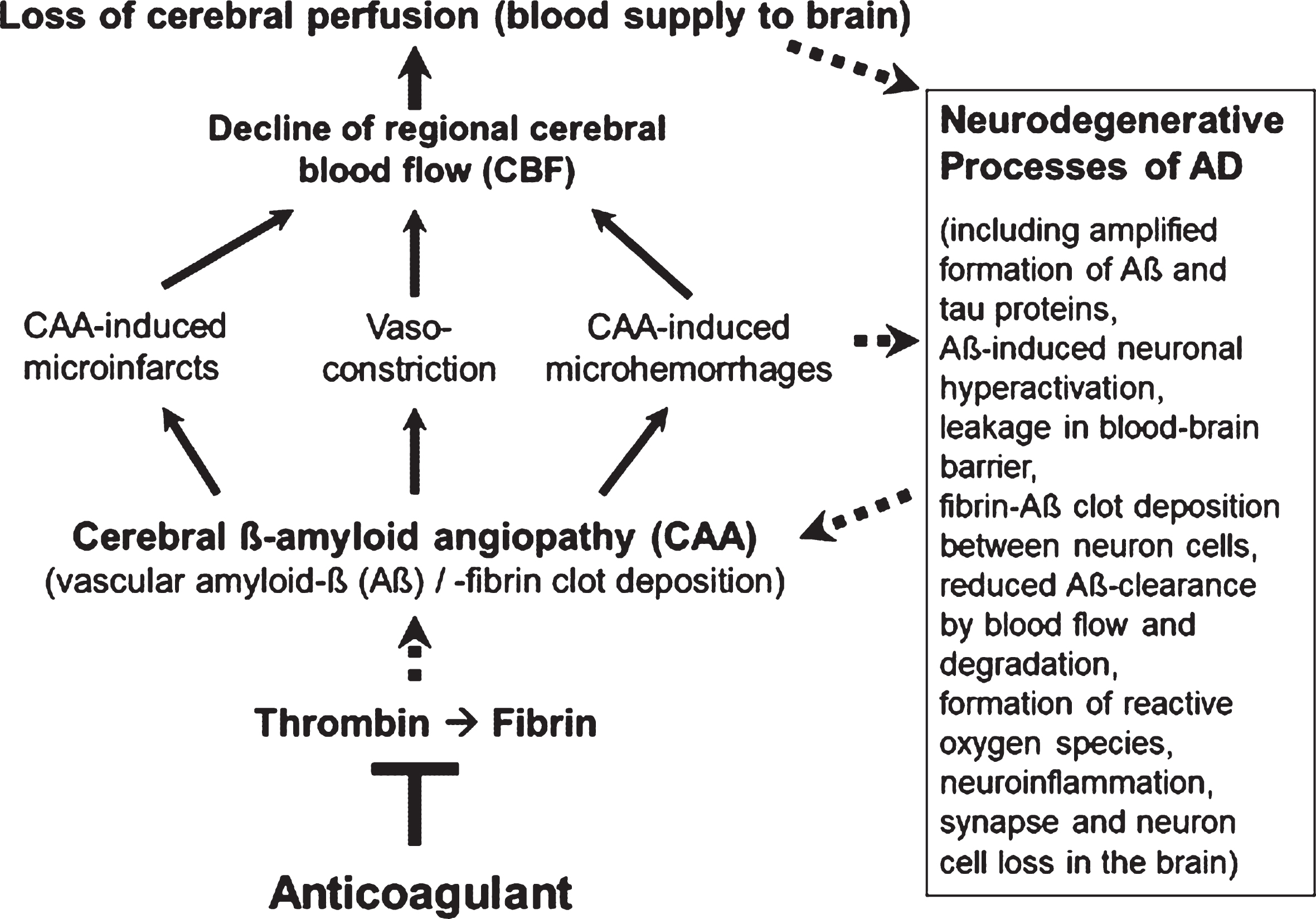
Vascular abnormalities, present in Alzheimer’s disease (AD), and proposed mechanism of action of direct oral anticoagulants (DOACs) for therapy. Typical of the early development of AD are pathological changes in the cerebral blood vessels, a condition, known as cerebral amyloid angiopathy (CAA). In the CAA, degradation-resistant fibrin clots containing amyloid-β-proteins (Aβ) deposit in and around cerebral blood vessels, leading to microvascular infarctions (occlusion), microhemorrhages and vasoconstriction of capillary vessels. Cerebral blood flow (CBF), mainly in neocortical and hippocampal areas, declines and brain perfusion and supply with oxygen and nutrients are lost. This amplifies neurodegenerative processes in the brain, exemplified by progressive Aβ and tau protein accumulation, reduced perivascular Aβ clearance, disruption of blood-brain barrier (BBB), neuroinflammation, and loss of synapses and neurons. In a kind of circulus vitiosus, vascular dysfunction and derived effects are steadily deteriorating. Anticoagulants inhibit thrombin and thus, the formation of fibrin at a central point of this process. Consequently, progressive fibrin-Aβ clot deposition in CAA and thrombin- and fibrin-induced inflammatory processes are blocked. Given full cerebral perfusion and reduced Aβ- and fibrin-accumulating and inflammatory milieu, it is proposed that anticoagulant treatment could be able to decrease vascular-driven progression in neurodegenerative and cognitive changes in AD. Indirect stimulatory effects are represented by dotted lines.
In addition, studies in AD mouse models and patients with genetic and sporadic AD revealed that, in parallel with Aβ, increasing amounts of the blood clotting-protein fibrin accumulate in the brain [57, 58]. Aβ was shown to bind to fibrinogen and fibrin, leading to fibrin deposits containing Aβ (fibrin-Aβ deposits). These were found in cerebral blood vessels in areas of CAA, and in brain parenchyma between neuron cells of AD mouse models and AD patients [57 –62] (Fig. 1). The accumulation of fibrinogen and fibrin in the brain parenchyma is caused by the fact that BBB becomes increasingly permeable during AD pathogenesis. Degeneration of pericyte cells in the wall of cerebral capillaries, leading to disrupted junctions between adjoining endothelial cells, was shown to contribute to BBB breakdown [63]. This allows that plasma proteins, such as thrombin and fibrin(ogen), can pass from the cerebral blood vessels into the brain parenchyma [54 , 63], leading to Aβ accumulation, microglial activation, synaptic dysfunction, and neuronal death [26]. Interaction between Aβ and fibrin(ogen) was found to alter the structure of the fibrin mesh. This forms persistent fibrin–Aβ clots with abnormal structure, which makes them resistant to clot-degrading enzymes in plasmin fibrinolysis [57 , 64]. Mutations in Aβ, which exist in patients with hereditary CAA, were found to promote the formation of cerebrovascular fibrin-Aβ clots in the pathogenesis [64]. These clots can hinder blood flow, promote inflammatory and degenerative changes in brain tissue, and can lead to the death of neurons [32]. Inhibitors, such as RU-505 and TDI-2760, which bind to Aβ and prevent interaction with fibrinogen and fibrin for clot formation, were shown to reduce vascular fibrin-Aβ clot deposition, vessel occlusion and microglia activation in the brain of AD mouse model. Ultimately, improved cognitive performance was observed [26, 65].
Furthermore, investigations in AD mouse models and AD patients have shown that Aβ activates the blood clotting factor FXII in the coagulation cascade for thrombin production (Fig. 2). This leads to an amplified formation of fibrin and fibrin–Aβ deposits in the vascular tissue and in brain parenchyma between neuron cells [66, 67]. Concomitantly, increased levels of proinflammatory thrombin and its precursor prothrombin were detected in the walls of cerebral microblood vessels and in brain parenchyma, particularly in neurons, glial cells and neural tau fibrillar deposits [67 –71]. Both thrombin and fibrin can cause intravascular and parenchymal inflammation in the brain [62, 70] (Fig. 2). Parenchymal fibrin was shown to activate microglia cells by binding the fibrin epitopeγ377–395 with the microglial receptor CR3, leading to inflammatory processes [72, 73]. Accordingly, blocking of the fibrin epitope by antibody reduced cerebral inflammatory and degenerative processes in AD mouse model [72]. It is also assumed that plasmin and plasminogen in the hemostatic system promote cerebral inflammatory processes [74]. Activation of the blood clotting factor FXII by Aβ also stimulated the production of the proinflammatory nonapeptide bradykinin via plasma prekallikrein and high molecular weight kininogen [67, 75] (Fig. 2). Recent studies in AD mouse model revealed a causal link between Aβ-induced FXII activation and neuroinflammatory, neurodegenerative and cognitive changes in AD [76].
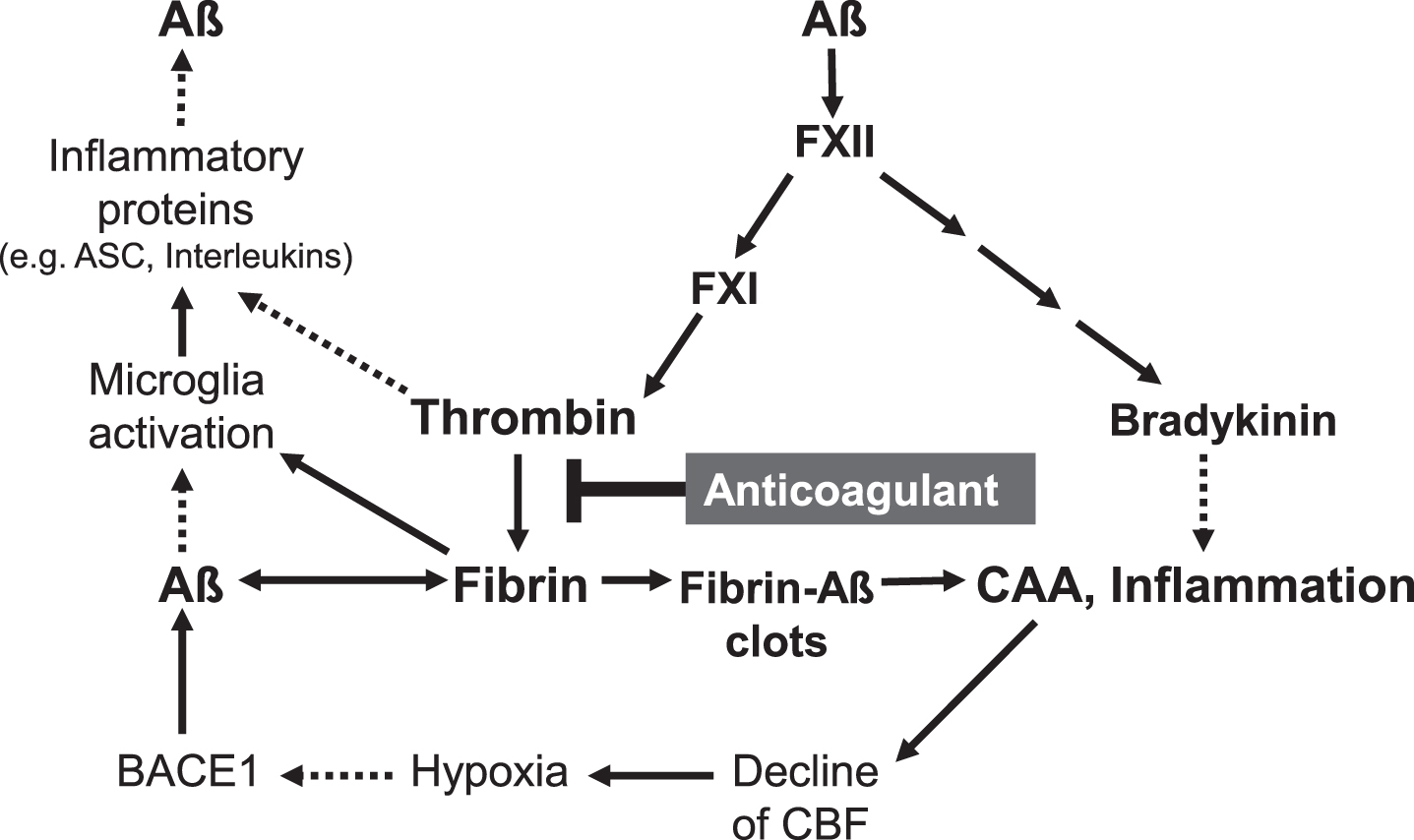
Key factors in cerebral amyloid angiopathy (CAA) and neuroinflammation are amyloid-β-proteins (Aβ), fibrin and thrombin (according to [26 , 66]). Aβ binds to fibrin(ogen) and forms degradation-resistant, Aβ-containing fibrin deposits (fibrin-Aβ clots), which are found in brain parenchyma between neuron cells and in cerebral blood vessels in areas of CAA. Aβ also activates the blood coagulation factor XII, leading to enhanced formation of proinflammatory thrombin and bradykinin, and microglia-activating fibrin. Aβ also triggers microglia activation. Activated microglia cells release, e.g., inflammatory interleukins and ASC (apoptosis-associated speck-like protein containing a CARD) protein complexes, which stimulate production and spread of cerebral Aβ and ultimately, amplify fibrin-Aβ deposition. CAA-induced decline of cerebral blood flow (CBF) and brain perfusion leads to tissue shortage of nutrients and oxygen (hypoxia), which stimulates β-secretase1 (BACE1) expression for amplified Aβ formation. Anticoagulant treatment intervenes in a central point of this catastrophic cascade. The drug inhibits thrombin and thus, the formation of fibrin. Progressive fibrin-Aβ clot deposition in CAA and brain parenchyma, thrombin- and fibrin-mediated inflammation and amplified Aβ production and derived neurodegenerative processes, contributing to AD, could be reduced by anticoagulant treatment. Indirect stimulatory effects are represented by dotted lines.
The results indicate a key vascular role of Aβ, fibrin, and thrombin in CAA with dramatic consequences for AD pathogenesis. Regional CBF in blood vessels declines considerably and therefore, brain perfusion and tissue supply with oxygen and nutrients suffer and neuroinflammatory and neurodegenerative processes are intensified (Figs. 1 and 2). The downward spiral of vascular and neurodegenerative changes is accelerated particularly by hypoxia-induced formation of Aβ and fibrin–Aβ clot deposits, reduced perivascular Aβ clearance, BBB disruption, and inflammation. Ultimately, these alterations can contribute to the loss of synapses and neuron cells and to cognitive decline.
NOVEL THERAPEUTIC APPROACH WITH ANTICOAGULANTS
The therapeutic approach is based on the hypothesis that anticoagulants affect the formation of key drivers in CAA and neuroinflammation, present in AD. By inhibiting thrombin for fibrin formation, anticoagulants can block accumulations of proinflammatory thrombin and microglia-activating fibrin, as well as the deposition of degradation-resistant, Aβ-containing fibrin clots. These fibrin-Aβ clots are detected in brain parenchyma and in cerebral blood vessels in areas of CAA, leading to disturbed cerebral blood flow. Consequently, anticoagulant treatment can counteract hypoperfusion and restricted supply of brain tissue with oxygen and nutrients. Concomitantly, hypoperfusion-enhanced neurodegenerative processes, such as progressive accumulation of Aβ via synthesis and reduced clearance, neuroinflammation, and synapse and neuron cell loss, can be reduced (Fig. 1). Given full cerebral perfusion and reduced Aβ- and fibrin-accumulating and inflammatory milieu, anticoagulants could be able to decrease vascular-driven progression in neurodegenerative and cognitive changes, present in AD, when treated early in the process.
Accordingly, previous studies with AD mouse models have shown that treatment with the indirect heparin-type thrombin inhibitor enoxaparin reduced cortical Aβ deposition [77, 78]. Furthermore, treatment with the direct thrombin inhibitor dabigatran etexilate decreased the amounts of vascular inflammatory proteins and ROS formation [70], as well as microglia activation [79]. Because of their inhibitory effect on vascular proinflammatory thrombin, it has been assumed that thrombin inhibitors are able to ameliorate symptoms in AD [80].
Disadvantages of a heparin infusion therapy, however, are that 1) fibrin-bound thrombin, a major stimulus of fibrin clot growth, is not inactivated, 2) non-specific plasma protein binding can affect anticoagulation unpredictably, and 3) thrombocytopenia can be triggered [4, 81]. On the other hand, oral anticoagulants, with respect of their application and profile of action, are an advantage for therapy of vascular dysfunction, present in AD. However, oral anticoagulants from VKA-type, such as warfarin, have undesirable side effects, which include bleeding complications, as well as vitamin K deficiency effects on important proteins of the vascular and nervous system [4, 82]. Much more suitable are the specific acting DOACs, which include the direct thrombin inhibitor dabigatran etexilate (Pradaxa®) and the blood clotting factor Xa-inhibitors rivaroxaban (Xarelto®), apixaban (Eliquis®), edoxaban (Lixiana®), and betrixaban (Bevyxxa®).
Among the trade DOACs, dabigatran etexilate could be preferred for a therapy of vascular dysfunction, present in AD, for the following reasons. Dabigatran etexilate is the prodrug form, which releases in vivo the active substance dabigatran. Dabigatran specifically inhibits the enzyme thrombin, which directly converts fibrinogen into insoluble fibrin at the end of the blood clotting cascade [83]. Compared to the VKA warfarin, dabigatran has a shorter half-life in humans [5] and was associated with reduced risk of ischemic stroke, intracranial hemorrhage by up to 66%, and death in elderly patients with atrial fibrillation [84, 85]. According to a U.S. Food and Administration (FDA) study, an incidence rate for intracranial hemorrhage per 1000 person-years of 3.3 (0.33%) was observed after treatment with dabigatran etexilate, whereas a rate of 9.6 (0.96%) was found for warfarin treatment [85]. Accordingly, dabigatran etexilate did not increase cerebral microbleeds in AD mouse model [86]. Due to the mechanism of action, dabigatran etexilate causes no vitamin K deficiency effects and nutritional interactions with anticoagulation [83]. The compound leads to effective, predictable and reliable anticoagulation [83]. Moreover, the trade antibody idarucizumab provides a specific antidote that neutralizes the effect of dabigatran within minutes and therefore, effectively counteracts bleeding risk [87]. Since recently, this option is also available for factor Xa-inhibiting DOACs by the reversal agent andexanet alfa, a modified recombinant inactive form of human factor Xa [88]. Nevertheless, use of dabigatran or other antithrombotic agents in AD patients that are more likely to bleed, given the association of CAA and vascular fragility, will need to be carefully evaluated for bleeding risk [89].
CONCLUSION
It is hypothesized that particularly anticoagulants from DOAC-type, such as the direct thrombin inhibitor dabigatran etexilate and indirect thrombin-affecting factor Xa inhibitors, are therapeutically suitable to achieve beneficial effects on cerebrovascular alterations, present in AD. Anticoagulants could counteract the formation of inflammatory thrombin and fibrin, and the deposition of degradation-resistant, Aβ-containing fibrin clots in cerebral blood vessels of CAA and in brain parenchyma. Applied early, therapeutically or prophylactically, dabigatran etexilate might be, for pharmacological reasons, preferentially used to ameliorate vascular dysfunction and inflammatory processes. Disturbance of cerebral blood flow, brain perfusion and supply with oxygen and nutrients, and increased progression of neurodegenerative processes, contributing to cognitive decline, could be reduced by anticoagulant treatment (Figs. 1 and 2). Since dabigatran etexilate is a clinically approved and over many years prescribed medicament with known safety profile, the therapeutic translation to this novel disease indication is probably faster and less expensively to achieve. However, it will be the task, first of preclinical research in AD mouse models, to study, if indeed this therapeutic approach is able to counteract effectively cerebrovascular dysfunction, present in AD pathogenesis. Interestingly, in the meantime, Cortes-Canteli and co-workers (2019) have reported results in AD mouse model, which are in line with the presented hypothesis [90]. Long-term treatment with dabigatran etexilate prevented memory decline, hypoperfusion, and fibrin deposition in the brain [90]. Concomitantly, the extent of Aβ plaques and oligomers, and neuroinflammatory activity, characterized by phagocytic microglia and infiltrated T cells, were reduced. In addition, BBB function was preserved, indicated by prevented astrogliosis and pericyte alterations. No hemorrhages or incidents of intracerebral bleeding were observed [90]. If these results will be confirmed by other investigations in AD mouse models, next, complex clinical testing of DOAC-type anticoagulants on therapeutic value in AD would be highly recommended. In clinical trials for testing the hypothesis, DOAC effects on early AD and CAA progression in patients can be studied by position emission tomography (PET) and magnetic resonance imaging (MRI) methods for visualizing brain biomarkers, such as plaques of Aβ, microbleeds, and CBF dynamics [14 , 91]. In addition, biochemical assays, preferentially for blood biomarkers, indicating neurodegenerative (e.g., Nfl [31]) and hemostatic changes (e.g., fibrin clot formation), as well as cognitive ability tests can complement the analysis. However, cost/benefit of the intended methods should be analyzed before study [91]. Nevertheless, every effort should be made, because currently no promising drug is recognizable on the horizon for the effective treatment of this terrible disease, with more than 40 million people worldwide thought to be afflicted [92, 93]. Of these, an estimated 5% develop symptoms usually well before the age of 65, due to familial (hereditary) AD predisposition (early-onset AD) [94].
Footnotes
ACKNOWLEDGMENTS
The author thanks particularly Prof. Dr. M. Jucker, University of Tübingen, Germany, and Prof. Dr. B. Wolozin, Boston University School of Medicine, USA, for valuable discussion, and his wife Regina for her great patience and support during more than 40 years at the side of a scientist.