
Abstract
Air pollution is a risk factor for cardiovascular and respiratory morbidity and mortality. A growing literature also links exposure to diverse air pollutants (e.g., nanoparticles, particulate matter, ozone, traffic-related air pollution) with brain health, including increased incidence of neurological and psychiatric disorders such as cognitive decline, dementia (including Alzheimer’s disease), anxiety, depression, and suicide. A critical gap in our understanding of adverse impacts of pollutants on the central nervous system (CNS) is the early initiating events triggered by pollutant inhalation that contribute to disease progression. Recent experimental evidence has shown that particulate matter and ozone, two common pollutants with differing characteristics and reactivity, can activate the hypothalamic-pituitary-adrenal (HPA) axis and release glucocorticoid stress hormones (cortisol in humans, corticosterone in rodents) as part of a neuroendocrine stress response. The brain is highly sensitive to stress: stress hormones affect cognition and mental health, and chronic stress can produce profound biochemical and structural changes in the brain. Chronic activation and/or dysfunction of the HPA axis also increases the burden on physiological stress response systems, conceptualized as allostatic load, and is a common pathway implicated in many diseases. The present paper provides an overview of how systemic stress-dependent biological responses common to particulate matter and ozone may provide insight into early CNS effects of pollutants, including links with oxidative, inflammatory, and metabolic processes. Evidence of pollutant effect modification by non-chemical stressors (e.g., socioeconomic position, psychosocial, noise), age (prenatal to elderly), and sex will also be reviewed in the context of susceptibility across the lifespan.
Keywords
INTRODUCTION
Studies conducted around the world have consistently shown that variation in air pollution levels is associated with cardiovascular and respiratory morbidity and mortality [1, 2]. Recently, health conditions associated with exposure to common air pollutants have broadened to include impacts on the brain such as anxiety, depression, cognitive deficits, and dementia. The societal implications are significant: neurological and mental health disorders globally represent the largest contributor to years lived with disability, and with an aging population the proportion of affected individuals is growing [3, 4]. Given the ubiquitous exposure of the population to air pollution, and the prevalence of psychiatric and neurological diseases, even small increases in relative risk translate to a substantial public health burden.
Despite compelling evidence that the brain is a target of air pollutants, and that inflammation and oxidative stress are common features of pollutant-induced disease processes, mechanisms linking pollutant inhalation to effects in the brain remain poorly understood [5 –7]. In particular, we lack insight into the early initiating events triggered by exposure to air pollutants that lead to disease processes in the brain. Elucidating mechanisms that directly link exposure to air pollutants with effects in the brain is a critical step toward addressing several key knowledge gaps. These include whether common or distinct processes underlie the relationship between exposure to air pollutants and diverse central nervous system (CNS) disorders (e.g., cognitive decline, dementia, depression), each of which has both common and distinct features and etiology. Understanding whether or not biological effects are specific to individual pollutants has implications for risk management and the targeting of key actors for regulatory action. Furthermore, with increased understanding of underlying mechanisms comes knowledge needed to identify those factors that contribute to susceptibility.
Recent work has shown that among the early biological responses triggered by exposure to air pollutants is a stress response that includes activation of the hypothalamic-pituitary-adrenal (HPA) axis and release of stress hormones. Dysregulation of the HPA axis is a feature of many disease processes common to both chronic stress and long-term exposure to air pollution, including cardiovascular disease, metabolic diseases such as type 2 diabetes, cognitive disorders, and depression [8]. Chronic stress and dysregulation of stress response systems have been proposed to contribute to both neurodegenerative diseases such as Alzheimer’s disease and to psychiatric disorders such as depression [9], making this mechanism an attractive target for investigation of mediators underlying CNS effects of air pollutants. The present paper examines how HPA axis dysfunction may contribute to CNS impacts associated with exposure to air pollutants. Effects of two common pollutants, ambient particulate matter and ozone, will be examined in relation to their differing physicochemical properties and mode of action. Recent evidence of stress axis activation by these pollutants will be summarized, and both the direct effects of stress axis activation on the CNS as well as indirect (systemic) consequences relevant to brain health will be reviewed. The potential contributions to brain disorders of converging biological pathways and their impact on cumulative physiological dysregulation, or allostatic load, will be discussed. Finally, the relevance of the stress response system to susceptibility will be examined by reviewing recent evidence of sex-dependent effect modification involving prior or co-exposure to non-chemical stressors across the life course.
PARTICULATE MATTER AND OZONE: DISTINCT PROPERTIES, OVERLAPPING HEALTH IMPACTS
Particulate matter (e.g., PM2.5 and PM10, particles with an aerodynamic diameter less than 2.5 and 10 μm, respectively) and ozone are currently considered to be the most important criteria pollutants with respect to morbidity and mortality. The Global Burden of Disease project attributed 4.2 million deaths and 103 million disability-adjusted life-years (essentially a measure of years of healthy living lost) to ambient particulate matter, with an additional 254,000 deaths and 4.1 million disability-adjusted life-years attributed to ozone [2]. As global mortality estimates have traditionally considered five specific causes of death (ischemic heart disease, lower respiratory infections, chronic obstructive pulmonary disease, lung cancer, and cerebrovascular disease), evidence of associations between air pollutants and reproductive (e.g., low birth weight), metabolic (e.g., diabetes), and neurological/mental health disorders suggests that the societal impact of air pollutants may actually be greater than currently appreciated. Indeed, recent work incorporating data from cohort studies spanning much of the range of global particulate levels yielded estimates of excess non-accidental deaths attributable to exposure to particulate matter that were significantly higher than previously recognized [10].
Air pollution is a complex mix of particles and gases that varies in relation to local and regional source contributions and atmospheric conditions. Particulate matter is itself a complex mixture that includes a number of toxic constituents such as transition metals, organics, sulfur, and black carbon derived from anthropogenic emissions as well as crustal sources. In vitro studies using cultured lung cell models have shown that particles compared on an equal mass basis can vary significantly in their cytotoxic and inflammatory potential in relation to spatial and temporal variations in contributions from traffic, industrial, and other urban sources [11 –13]. Although challenging to study at the population level, there is evidence that such differences in composition contribute to spatial variation in health impacts [14, 15]. However, while characteristics such as surface area, transition metals, and most recently oxidative potential have, among others, been identified as potential drivers for health effects of particulate matter [16], to date, mass concentration of a given particle size fraction continues to be the key metric used for regulatory action.
Even less is known about drivers of CNS effects. Because of their size and properties, considerable experimental work has focused on potential translocation of nanoparticles to the systemic circulation and brain, and on pathological effects produced in the brain following chronic exposure to particles, resulting in important findings supporting the plausibility of pollutant effects on brain health [7 , 17–19]. Depending on size and chemical composition, particulate matter or soluble constituents may translocate from the lungs to the systemic circulation, or migrate via olfactory transport, and directly interact with extrapulmonary cells and tissues including the brain [18, 20]. However, pollutants need not physically reach the brain, nor must there be chronic exposure, to provoke effects. A number of experimental studies (e.g., [21 –24]) have identified structural, functional, biochemical, and transcriptional alterations in the brain following both acute and repeated exposure to the highly-reactive gas ozone. Because of its reactivity, ozone is consumed within the lungs through reactions with lipids, proteins, and antioxidants present in the airway surface lining, and extrapulmonary ozone toxicity is thought to be mediated by secondary reactive products and other biological mediators released into the systemic circulation [25, 26]. Particulate matter, too, may impact the brain through indirect processes, such as through effects secondary to peripheral oxidative stress and inflammation, or to stimulation of pulmonary neuronal afferents, as has been proposed to explain cardiovascular impacts [1, 7].
Despite their differing properties and potential routes of exposure, both particulate matter and ozone have been associated with a variety of CNS impacts. These include impaired cognitive performance [27 –29], dementia [30, 31], anxiety and depression [32 –35], and suicide [36 –38]. It should be noted that there is considerable variability in the epidemiologic literature; a more comprehensive overview of associations between particulate matter or ozone and neurological/mental health outcomes is provided in several recent reviews [5 , 40]. In experimental models, repeated exposure to air pollutants including ambient particulate matter and ozone has been shown to increase oxidative stress and cytokine production in the brain, with evidence also of microglial activation and impacts on neurotransmitters, neuronal morphology, markers of neurodegenerative disease, altered cognition, and depressive-like behaviors (e.g., [21 , 41–43]). Collectively, epidemiological and toxicological studies have found that both particulate and gaseous pollutants, despite distinct physicochemical characteristics and biological reactivity, can impact brain health. Notwithstanding advances in our understanding that exposure to ambient pollutants can adversely impact the CNS, a critical knowledge gap remains the initiating mechanism(s) through which exposure to pollutants leads to effects in the brain.
INITIAL EFFECTS OF PARTICULATE MATTER AND OZONE: EVIDENCE OF A COMMON MEDIATOR
The physicochemical differences between particulate matter and ozone provide a useful contrast to investigate mechanisms underlying extrapulmonary effects of air pollutants. Unlike effects of ambient particles, which could be due to direct effects of the nanosized fraction or soluble constituents on target tissues, or to secondary effects from signaling via the systemic circulation or nervous inputs, extrapulmonary effects of ozone are attributable to secondary mediators. Extrapulmonary effects that are common to both pollutants could therefore provide evidence of a common secondary mediator or mediators, while distinct effects provide insight into pollutant-specific processes.
To investigate systemic impacts of inhaled pollutants, we exploited this contrast by comparing gene expression profiles in rats exposed to particulate matter, ozone, or both pollutants (Fig. 1A) [44]. We measured the expression of a set of genes representing a number of biological pathways in the lungs, heart, liver, kidney, spleen, cerebral hemisphere, and pituitary gland. We reasoned that examination of the pattern of response immediately after a single acute (4 h) exposure would allow assessment of initial pollutant effects not significantly influenced by subsequent cellular changes that accompany disease progression observed in repeated or chronic exposure models. Effects of exposure to particulate matter or ozone were observed in every organ, including robust effects in the cerebral hemisphere and pituitary. Remarkably, despite contrasting effects in the lungs (e.g., strong induction of interleukin-6 by ozone, and of xenobiotic response genes by particles), a subset of genes that included inflammatory, antioxidant, and stress-responsive factors exhibited a similar pattern of response to the pollutants across most organs. The pronounced response observed in the pituitary, coupled with responses common to all tissues assessed, led us to hypothesize endocrine involvement. Subsequent analyses confirmed that both particulate matter and ozone activated the HPA axis and provoked the release of the stress hormones adrenocorticotrophic hormone (ACTH) and corticosterone (Fig. 1B) [44].
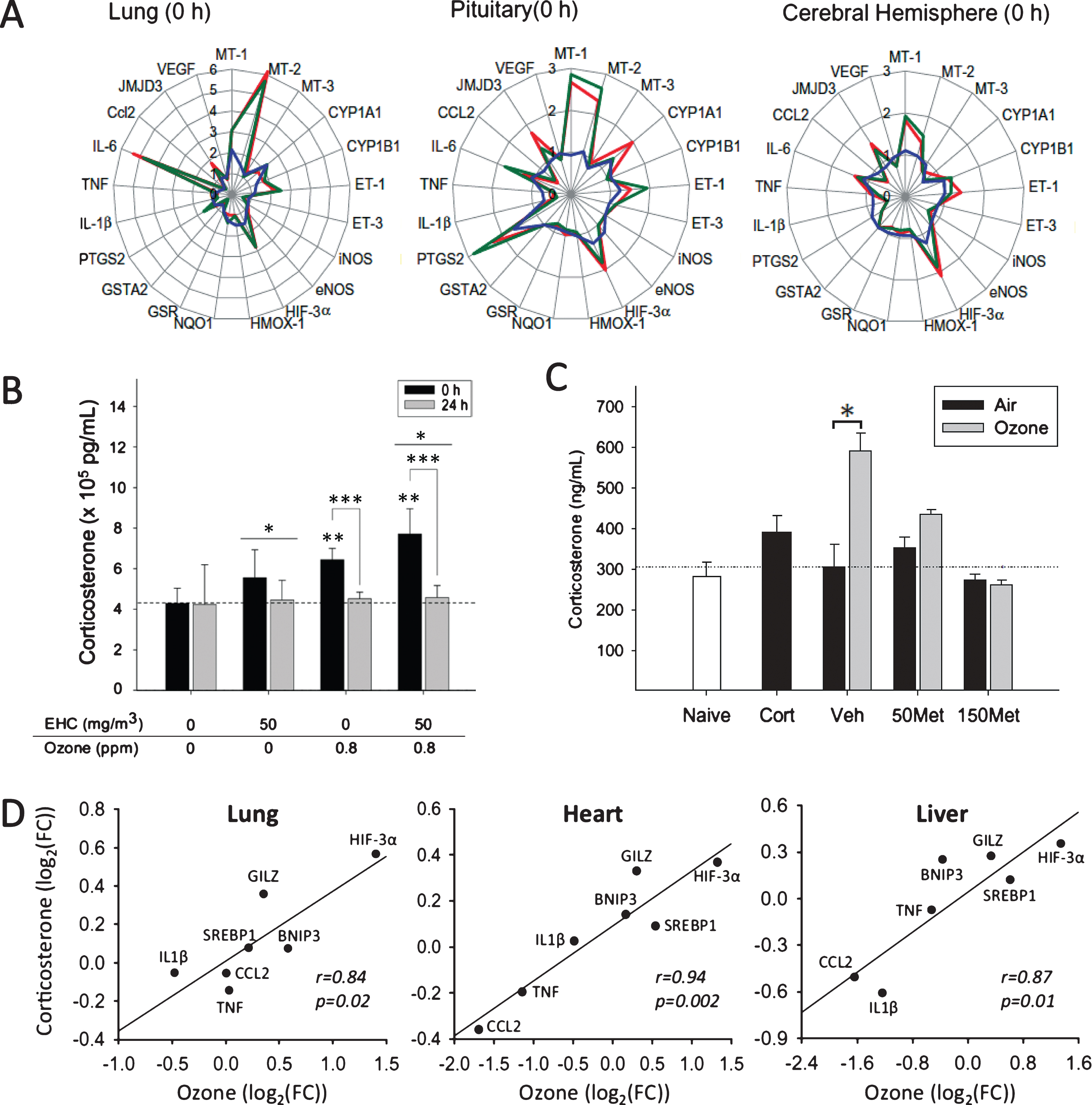
Systemic effects of particulate matter and ozone include activation of the hypothalamic-pituitary-adrenal (HPA) stress axis. A) Gene expression profiles (fold-change relative to air-exposed controls) were mapped across a variety of tissues in Fischer rats exposed by nose-only exposure for 4 h to particulate matter (blue), ozone (red), or particulate matter and ozone (green). B) Both particulate matter (EHC) and ozone increase plasma corticosterone (*significant particle effect; **significant ozone effect; ***significant time effect). C) Treatment with the drug metyrapone (50 mg/kg, 150 mg/kg Met) blocked the ozone-induced increase in corticosterone (n = 5/group). Naïve, rats not exposed to experimental paradigm; Cort, rats administered exogenous corticosterone and exposed to air. D) Administration of corticosterone (10 mg/kg) reproduced effects of ozone exposure (FC, fold-change). Figures reproduced or adapted from references 44 (A, B) and 54 (C, D).
THE HPA AXIS: AIR POLLUTANTS AS STRESSORS
Together with the sympathetic nervous system, the HPA axis plays a pivotal role in coordinating the response to stressors. Stressor exposure triggers hypothalamic production of corticotrophin-releasing hormone; this signals the pituitary to synthesize ACTH, which is released into the systemic circulation. Upon reaching the adrenal glands, ACTH stimulates de novo synthesis of glucocorticoids (primarily cortisol in humans, corticosterone in rodents) that are in turn released into circulation. Glucocorticoids act primarily through specific receptors to exert profound effects on a variety of processes that include glucose and lipid metabolism, immune response, and adipocyte differentiation, as well as interacting with other endocrine systems [45]. Depending on the intensity and duration of exposure, glucocorticoids can both stimulate and inhibit immune responses [46, 47]. Glucocorticoid production is shut down by means of a negative feedback mechanism. Acute activation of the HPA axis is essential for survival, providing the body with the means to respond to acute stressors including mobilizing and replenishing energy reserves and regulating immune responses. However, chronic activation of the HPA axis produces various deleterious consequences: both chronic stress and excess glucocorticoids are associated with increased risk of cardiovascular disease, metabolic dysfunction, depression, and reduced cognitive function [48]. There is a substantial overlap between stress-related diseases and diseases associated with exposure to air pollutants [8].
Recent experimental work has demonstrated that air pollutants can act as stressors and elicit endocrine stress responses. Exposure of rats to concentrated ambient particles, with and without ovalbumin-induced allergic disease, activated stress centers in the brain and increased circulating levels of corticosterone [49]. Short-term exposure of rats to ozone and particulate matter altered expression of pituitary endothelin-1 [23], a regulator of pituitary hormone secretion [50]. Ozone exposure activated stress centers in the brain of adult rats, with the pattern of activation suggesting signaling via the vagus nerve [51]. Repeated exposure of seven-week old female Wistar rats to ozone (0.12 ppm, 6 h/day for 15 d) resulted in higher plasma corticosterone that coincided with behavioral changes [52]. Acute exposure to particulate matter or ozone increased plasma levels of the stress hormones ACTH and corticosterone and altered expression across multiple tissues in Fischer rats, confirming that a single exposure to either particulate or gaseous pollutants can activate the HPA axis and produce common (and additive) systemic effects [44]. Short-term exposure to ozone increased epinephrine levels in Brown Norway rats [53], consistent with a response of the sympathetic nervous system as part of the stress response. Blocking the ozone-induced increase of corticosterone using the drug metyrapone increased inflammatory signaling in the lungs and circulation, consistent with immunosuppressive action of glucocorticoids, and prevented effects of ozone on metabolic and inflammatory factor expression in several organs [54]. Effects of ozone were reproduced by administration of corticosterone (Fig. 1C, D), confirming a role for the HPA axis in mediating and modifying local and systemic effects of ozone exposure [54]. More recently, evidence of adrenal stress hormone involvement was further substantiated in a study that showed that administration of adrenergic and glucocorticoid receptor antagonists prior to and concurrent with exposure modified the pulmonary response to ozone [55]. The relevance of these observations to humans was confirmed in a panel study that showed that controlled exposure to 0.2 ppm ozone increased plasma stress hormone levels [56]. Furthermore, reduction of indoor particulate matter levels using a particle filter was associated with a drop in cortisol levels in a randomized double-blind crossover design study [57]. Collectively, these studies show that exposure to gaseous and particulate pollutants can elicit neuroendocrine stress responses.
DIRECT EFFECTS OF STRESS AXIS DYSREGULATION ON THE BRAIN
The demonstration that air pollutants can activate the HPA stress axis has important implications with respect to potential direct and indirect effects on the brain (Fig. 2). Effects of stress and stress hormones on the brain have been investigated for decades, leading to insights into impacts of acute and chronic stressors on molecular signaling, neuronal structure, and brain plasticity [58, 59]. Both acute and chronic stress produce neurochemical and structural changes in the brain that have consequences for neurodevelopmental processes as well as healthy aging [60]. Cortisol plays a central role in regulating fetal brain development, including the regulation of neurotrophic factors such as serotonin [61]. Stress and elevated glucocorticoid levels are implicated in prevalent brain disorders that include depression and dementia [48 , 62–64]. Among older adults, individuals with progressively higher basal cortisol levels over several years were found to have greater memory impairment and lower hippocampal volume than individuals with lower cortisol levels [65]. Interindividual differences in HPA axis reactivity explain the relationship between serotonin transporter gene single nucleotide polymorphisms and susceptibility to depression [66], suggesting that differences in stress response may also be important modifiers of the effect of chronic stressors on mental health.
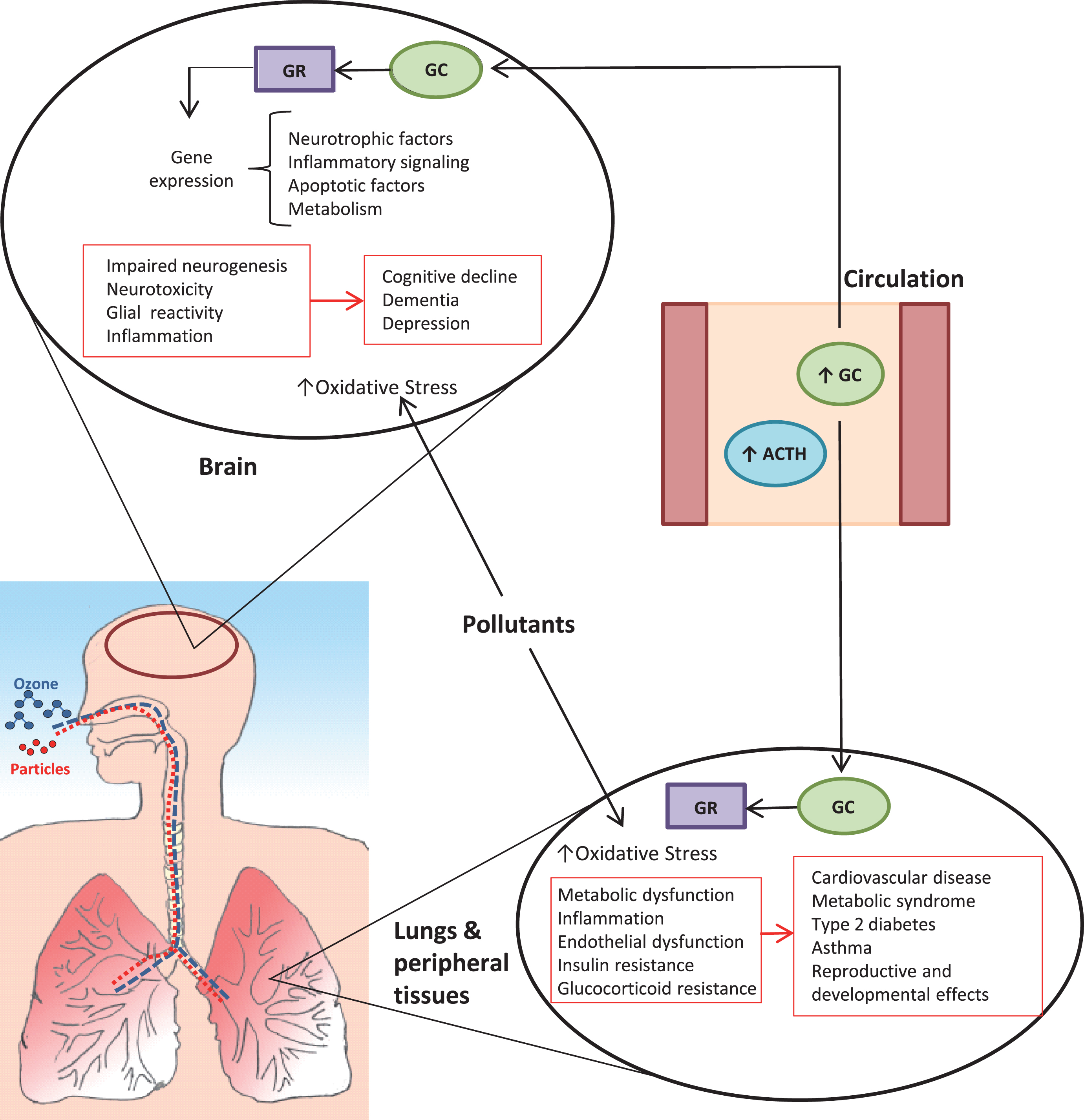
Proposed direct and systemic impacts of pollutant-induced stress axis activation on the brain. Both particulate matter and ozone trigger a stress response, resulting in pituitary release of adrenocorticotrophic hormone (ACTH), which in turn signals the adrenal glands to increase production of glucocorticoids (GC; primarily cortisol in humans, corticosterone in rodents). Glucocorticoids bind to receptors (GR) to regulate processes that include glucose and lipid homeostasis, immune/inflammatory function, and responses to other hormones. The stress hormone release coincides with and contributes to changes in blood mediators (e.g., cytokines, metabolic factors, reactive products) that have systemic impacts, including effects on the brain, lungs, and peripheral tissues. Chronic activation and dysregulation of the HPA axis is associated with a variety of adverse effects that include neurotoxicity, sensitization to other insults including oxidative stress, and impaired control of inflammatory processes. Collectively, these effects interact with individual susceptibility to contribute to disease processes that manifest systemically and in the brain.
The brain is highly sensitive to glucocorticoids. Much of our understanding of the effects of glucocorticoids on the brain were founded upon original studies examining effects in the hippocampus, which were then extended to other regions including the amygdala and prefrontal cortex [58]. Under the glucocorticoid-cascade hypothesis (now called the neurotoxicity hypothesis), effects of cumulative exposure to elevated glucocorticoid levels can lead to hippocampal atrophy [67]. Stress is a strong negative regulator of hippocampal neurogenesis, thought to be a key cause of morphological and functional deficits observed in depression [68]. Because the hippocampus contributes to regulation of the HPA axis, this effect can contribute to HPA axis dysregulation, a feed-forward effect that may eventually impair cognition [67, 69].
Both particulate matter and ozone alter the expression of glucocorticoid-regulated genes in the brain of exposed rats ([44] and unpublished observations), consistent with biologically-effective increases in cerebral glucocorticoid concentrations following pollutant exposure. While it remains to be determined to what extent stress axis dysfunction is implicated in adverse CNS impacts of air pollutants, there is considerable experimental evidence to support a direct causal role for glucocorticoids and HPA axis dysfunction in brain pathologies. Repeated exposure of rats to exogenous corticosterone impaired memory and accelerated hippocampal neuronal loss and glial reactivity, while adrenalectomy alleviated these characteristics of brain aging [70, 71]. Glucocorticoid hypersecretion is associated with reduced hippocampal volume and memory impairments in rats [72], showing that individual differences in glucocorticoid levels may be relevant to brain health. Depending on dose, duration, and brain region, glucocorticoids appear to differentially impact proliferation of oligodendrocyte progenitors [73, 74], and microstructural changes indicative of loss of integrity and demyelination have been observed in the white matter of patients with Cushing Syndrome [75]. In addition to their well-established anti-inflammatory role, glucocorticoids can exert early permissive immunostimulative effects in the periphery, as well as proinflammatory effects during the immune response to injury in the CNS, that appear to depend upon level, timing, and duration of exposure [47]. Chronic administration of glucocorticoids has been shown to increase amyloid-β prevalence in the brain of aged macaques, possibly by impairing expression of insulin-degrading enzyme, a candidate protease for clearance of amyloid-β peptides [76]. 11β-hydroxysteroid dehydrogenase 1 –/– mice, which lack a key enzyme responsible for tissue-specific conversion of glucocorticoid precursors to the active form, are protected from the cognitive decline that affects aged wild-type mice [77], emphasizing the importance of tissue glucocorticoid levels in mediating effects. Repeated exposure to corticosterone is used as a model of depression [78], and subchronic treatment with a glucocorticoid receptor inhibitor prevented the cognitive decline observed in a transgenic mouse model of Alzheimer’s disease [79]. Collectively, these and other studies establish a causal link between dysregulated glucocorticoids and impacts on cognition, neurodegeneration, and depression.
STRESS-DEPENDENT SYSTEMIC INVOLVEMENT IN CNS DYSFUNCTION
Systemic effects of pollutant exposure, like systemic effects of chronic stress, may also contribute to impacts on the brain. Major systems and factors that contribute to an integrated stress response include the sympathetic nervous system, immune system (including actions of inflammatory cytokines), and metabolic factors. Chronic elevation of glucocorticoids, as seen in Cushing Syndrome, is associated with hyperglycemia, impaired immune function, hypertension, obesity, and depression [45, 80]. The hypercortilism and inflammation seen in stress-related diseases such as cardiovascular disease, metabolic syndrome, and depression have been proposed to result from glucocorticoid resistance [80]. Co-morbidity of metabolic and neurological/neurobehavioral disorders is well-established, and is thought to relate at least in part to common underlying dysfunction of the HPA axis [48 , 81–83].
The brain is highly sensitive to metabolic disturbance, oxidative stress, and inflammatory stimuli. Glucocorticoids exert well-known impacts on metabolic systems, including antagonizing insulin signaling and regulating glucose and fatty acid homeostasis [84, 85]. Insulin signaling is implicated in neuronal and cognitive function [86], and insulin resistance is associated with increased risk for Alzheimer’s disease [87]. Insulin-degrading enzyme is implicated in the regulation of amyloid-β levels, with knockout mice exhibiting increased cerebral accumulation of amyloid-β, hyperinsulinemia, and glucose intolerance [88]. Reduced glucose tolerance has been associated with poor memory performance and hippocampal atrophy [89], and insulin resistance was associated with reduced cerebral glucose uptake and increased risk of Alzheimer’s disease [90]. Experimental models have shown that repeated exposure to corticosterone produces a model of depression [78], and when combined with a high-fat diet produces a model of type II diabetes [91], implicating glucocorticoids in metabolic and neurobehavioral disorders.
There is abundant epidemiological and experimental data to show that exposure to air pollutants can lead to dysregulation of metabolic and inflammatory processes [92]. For example, mice exposed chronically to concentrated ambient particles and a high fat diet developed insulin resistance, systemic inflammation, and increased visceral fat deposition [93]. Acute exposure to ozone produced glucose intolerance in Brown Norway rats [53], and in mice produced insulin resistance partially reversed by antioxidant treatment [94]. There is currently limited data to show to what extent the stress axis is involved in such effects, particularly in chronic exposure models, as data to date come from short-term exposure studies. Pharmacological intervention with metyrapone, a drug that blocks corticosterone synthesis, prevented a subset of acute ozone-dependent metabolic effects in male Fischer rats and enhanced the release of some cytokines into the circulation after ozone exposure [54]. Adrenalectomy alleviated acute ozone-induced decreases in glucose tolerance in Wistar-Kyoto rats [95], consistent with the involvement of adrenal hormones. Ozone-dependent changes in the response of metabolic and endocrine factors to glucose challenge were reproduced by administration of corticosterone, supporting a role for glucocorticoids in mediating pollutant effects [96]. Given the co-morbidity of metabolic and neurological and neurobehavioral disorders and the sensitivity of the brain to metabolic and inflammatory stimuli, such changes may have relevance to impacts of pollutants on the brain.
CONVERGING PATHWAYS: OXIDATIVE STRESS, INFLAMMATION, AND THE NEUROENDOCRINE STRESS RESPONSE
The diverse health impacts now associated with exposure to air pollution suggest involvement of a number of processes that manifest according to the specific makeup of the pollutant mix and interindividual differences in susceptibility. Stress axis dysfunction is unlikely to be the only factor involved. Oxidative stress and inflammation have long been considered important features of disease processes initiated by pollutants, including effects in the brain, where lipid peroxidation, microglial activation, and increased levels of inflammatory cytokines have been observed in experimental models following both acute and chronic exposure [97, 98]. These, along with impacts on neurotransmitter systems and changes of neuronal structure and function, have been proposed as prominent mechanisms underlying CNS impacts of pollutant exposure [6, 7].
Recent human data support involvement of oxidative stress and inflammatory pathways in a variety of pollutant effects relevant to brain health. For example, associations between prenatal exposure to ambient pollutants and impaired cognitive development were strongest in the offspring of mothers who reported low fruit and vegetable consumption, important sources of antioxidants [99]. Mitochondrial DNA copy number, which increases in response to environmental demands and oxidative damage, has been associated with exposure to pollutants in utero and in later life [100, 101]. Although the physiological consequences are not entirely clear, mitochondrial damage is associated with a variety of disorders including neurodegenerative diseases [102]. Black carbon exposure averaged over a year was associated with reduced cognitive function, with stronger effects observed in individuals with longer leukocyte telomeres, a characteristic associated with the capacity to mount a stronger systemic inflammatory response [103]. In addition to this potential role as an effect modifier, telomere length may also reflect oxidative and inflammatory stresses linked to exposures, with long-term exposures to a range of occupational and environmental toxicants generally associated with shorter leukocyte telomere length [104]. This emerging literature supports a role for air pollution in contributing to fetal programming and to accelerated aging, at least in part through cumulative effects of oxidative and inflammatory stresses.
Basal glucocorticoid levels and pollutant-dependent regulation of the HPA axis may contribute to such effects. Rodent studies have shown that the hippocampus, in addition to being highly sensitive to glucocorticoids, appears also to be particularly sensitive to the oxidative stress caused by ozone exposure [105, 106], and exhibits inflammatory signaling, impaired neurogenesis, and altered neuronal morphology following long-term exposure to concentrated ambient particles or traffic-related air pollutants [42 , 108]. In addition to exerting neurotoxic effects and impairing neurogenesis, chronic stress and elevated glucocorticoids can prime microglial proinflammatory responses to subsequent insults [109, 110]. Like oxidative stress, psychological stress and elevated glucocorticoid levels are associated with accelerated telomere shortening [111 –113]. Glucocorticoids also regulate mitochondrial function, correlating with neuroprotection at low levels and neurotoxicity at high levels [114]. As air pollutants may act through diverse pathways, such combined effects may render specific brain structures vulnerable to air pollutants, depending on individual host susceptibility and exposure to other stressors.
ALLOSTATIC LOAD: CUMULATIVE PHYSIOLOGICAL DYSFUNCTION FROM CHRONIC STRESSOR EXPOSURE
Adaptation to stressors requires an integrated response that is essential for survival, but is not without cost. Diseases associated with chronic stress, as with chronic exposure to air pollution, are characterized by common underlying processes that include dysregulation of endocrine, inflammatory, and metabolic systems. If air pollutants act as stressors through impacts on the HPA axis, considering the combined and cumulative effects of multiple stressors and innate differences in stress reactivity may be important to understand conditions that contribute to susceptibility. Key concepts that may help to explain how the cumulative burden imposed by exposure to stressors increases the likelihood of morbidity and mortality include allostasis, which describes the process by which the body responds to exposure to stressors, and allostatic load, which describes the wear and tear on the body as it responds to stressors [115]. In essence, allostatic load represents the physiological consequences of prolonged exposure to stressors interacting with individual host sensitivity. While stress response systems have evolved to respond to acute stressors by providing a survival advantage, chronic activation imposes a burden that may alter the capacity to respond to new challenges, increasing vulnerability to new stressors and contributing to disease processes (Fig. 3). This, coupled with interindividual variability in stress responses, may explain why small changes in the levels of a stressor that is tolerated in some individuals can, in others, produce an adverse effect.
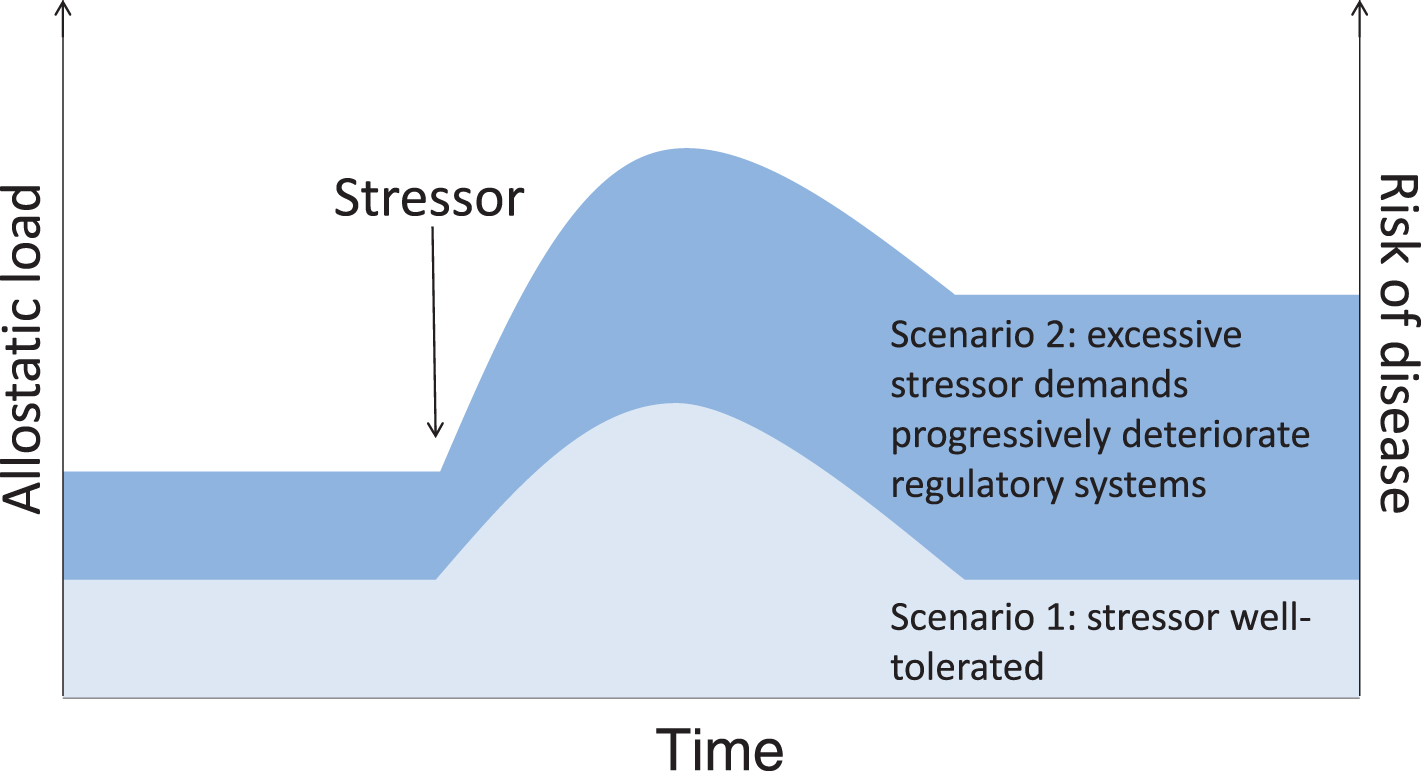
Allostatic load scenarios. Both intrinsic (e.g., stress reactivity, resilience, existing disease states) and extrinsic (e.g., prior and concurrent exposure to other psychosocial, physical, or chemical stressors) factors will interact to determine to what extent stressor exposure contributes to physiological dysfunction, or allostatic load, and increases the risk of disease.
Efforts to operationalize the allostatic load concept using composite indices that incorporate a set of biological measures covering physiological processes dysregulated by chronic stress (e.g., cortisol, epinephrine, cholesterol, glycated hemoglobin, blood pressure, waist-to-hip ratio, C-reactive protein) have shown its utility in predicting mortality, as well as physical and cognitive decline [116]. For example, higher baseline allostatic load scores in men and women aged 70–79 were associated with a steeper decline in cognitive function over a seven year follow-up period [117]. In a large cross-sectional study, higher allostatic load scores were associated with poorer working memory in adults aged 20–59 [118]. Allostatic load during childhood was associated with poorer working memory in young adults [119]. Such relationships may have relevance in interpreting associations between pollutants and health outcomes. For example, long-term exposure to air pollutants was associated with impaired performance on mathematical and verbal tests, with effects of pollutant exposure on verbal skills most evident with increasing age and lower education [27].
The concept of allostatic load provides insight into social gradients in health that relate to exposure to chronic stressors, including air pollution. It has been recognized for some time that socioeconomic position, in addition to being a potential confounder of associations between exposure to air pollutants and health outcomes, may also be an effect modifier. O’Neill and colleagues identified three mechanisms that may explain the increased adverse effects on health associated with air pollution among individuals of lower socioeconomic position, namely: 1) susceptibility directly related to socioeconomic position, such as through higher levels of psychosocial stress, limited access to health care, or increased likelihood of living in lower quality housing and associated greater exposure to stressors such as noise, crowding, violence, allergens, and other pollutants; 2) higher prevalence of health conditions and behaviors that increase susceptibility such as diabetes and smoking; and 3) more frequent or more intense exposures to air pollution [120]. Stress may be common to all three. If air pollution acts as a chronic stressor, contributing to allostatic load and accelerating the progression of stress-associated diseases, gradients in exposure to stressors and individual differences in sensitivity and behavior may act as important modifiers of health impacts. Factors associated with resilience may also modify the response to pollutants, as suggested by evidence that exposure to natural environments (e.g., green and blue spaces) is associated with protective effects, including improvements in allostatic load measures, cardiovascular and mental health outcomes, and risk of cause-specific mortality [121 –123].
VULNERABILITY: INTERACTIONS WITH SEX, AGE, AND NON-CHEMICAL STRESSORS
Differences in the magnitude and nature of effects of air pollutants on health are thought to depend both on subject-specific traits (such as age, sex, existing disease, genetic variation, behaviors) and contextual characteristics (such as socioeconomic position, concurrent environmental exposures), but the specific factors and mechanisms that define susceptibility remain uncertain [120, 124]. In evaluating health risks associated with exposures, cumulative risk assessment initiatives have emphasized the importance of considering non-chemical stressors alongside chemicals, as well as interactions with intrinsic factors such as age and sex [125 –128]. Such interactions may be important in the context of stress because effects of stressors will vary across the lifespan. For example, with respect to effects on the brain, stress may interfere with developmental programming during gestation and early childhood, contribute to depression in adults, and accelerate cognitive decline in the elderly [60]. Moreover, stress hormone responses to a standardized acute psychosocial stressor (the Trier Social Stress test) have been found to vary in relation to age and sex [129]. Accordingly, consideration of the stress context during specific life stages in each sex may be needed to more fully understand effects of pollutants and the modifying role of intrinsic and extrinsic factors (Fig. 4).
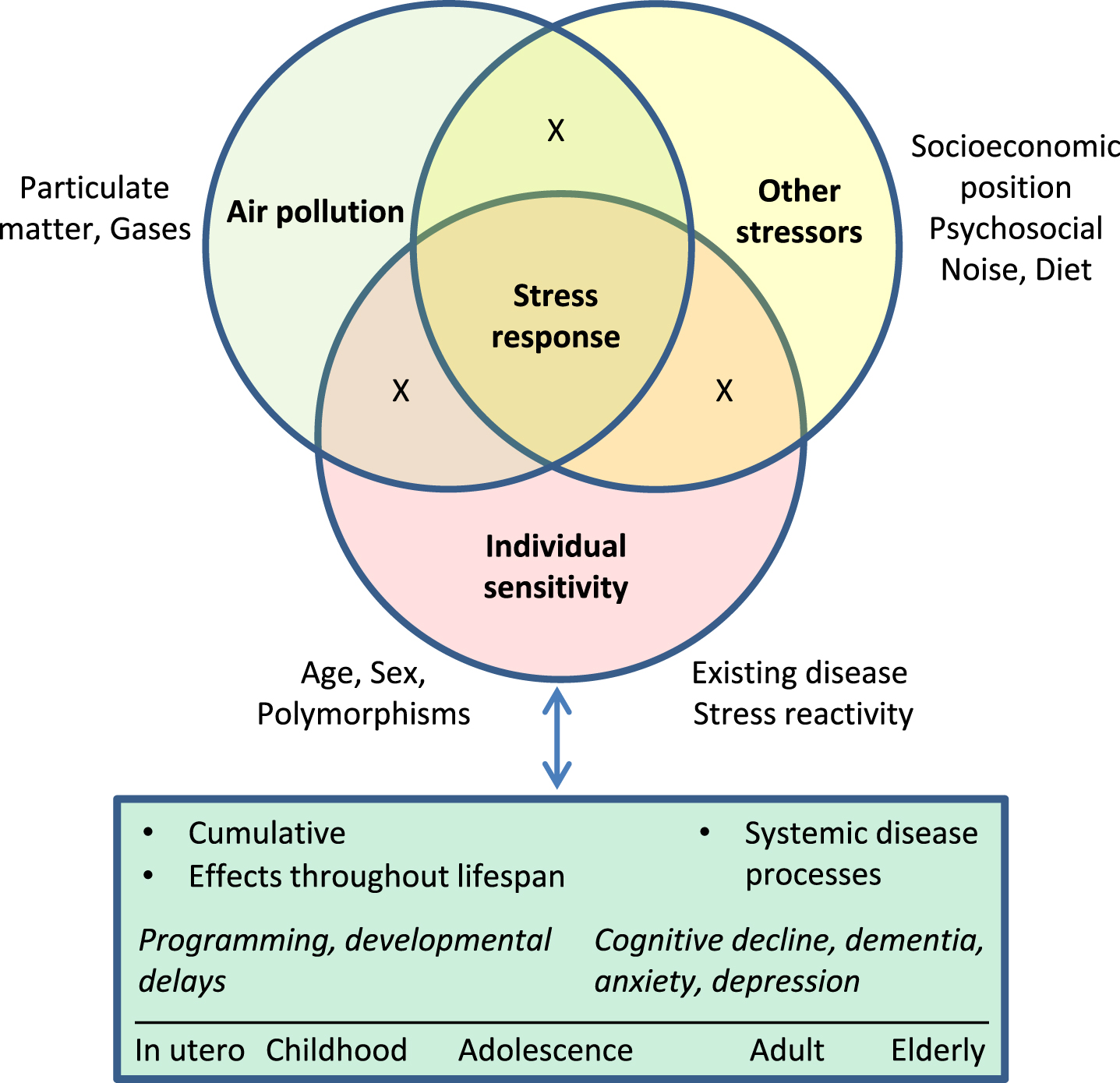
Stress response as an integrator of effects of chemical and non-chemical stressors and individual sensitivity on brain health across the life course. Exposure to air pollution occurs in the context of other exposures, including stressors associated with socioeconomic position such crowding, crime, noise, and other hazards. Deleterious effects of exposure to chronic stressors may accumulate across the life course, and in turn may further increase vulnerability to subsequent exposures. Effects include impacts on fetal development, programming of future stress reactivity and disease susceptibility, and cumulative dysregulation of systemic neuroendocrine, cardiovascular, inflammatory, and metabolic processes that collectively contribute to disease. “X” represents interactions among factors.
In this vein, a number of recent studies have directly examined the link between the stress context and pollutant effect modification, including examination of specific vulnerability periods in each sex. Higher PM2.5 exposure during the first trimester was associated with increased risk of wheeze among children with mothers who reported high prenatal stress [130]. Prenatal nitrate exposure at 7–19 and 33–40 weeks gestation was associated with asthma in boys exposed to high prenatal stress [131]. Combined exposure to PM2.5 and maternal stress was associated with mitochondrial DNA copy number in cord blood and placenta that also differed in a sex-specific manner [132]. Prenatal exposure to PM2.5 was associated with childhood asthma, with stronger effects observed in boys exposed prenatally to maternal stress [133]. Stronger associations between indicators of traffic-related air pollution (NO2 and/or oxides of nitrogen) and asthma incidence have been reported for children exposed to community violence [134] or living with higher parental stress [135], suggesting that heightened stress levels may exacerbate respiratory effects of pollutant exposure. Chronic stress was also associated with increased vulnerability to asthma exacerbations in children exposed to relatively low levels of traffic-related pollutants, an interaction not observed at higher pollutant levels [136]. Co-exposure to psychosocial stress was associated with greater risk of decreased lung function from exposure to traffic-related air pollutants in children [137] and adolescents [138]. In adults, stronger associations between PM2.5 and systolic blood pressure were found in individuals who reported high psychosocial stress assessed using a stress index that included neighborhood stress, acute life events, family caregiving stress, financial vulnerability, and unfair treatment [139], an association not found in a previous study that did not consider multiple sources and types of stress [140]. Collectively, these studies suggest that prior and concurrent exposures to non-chemical stressors exacerbate effects of air pollutants on respiratory and cardiovascular disease across life stages, including through prenatal programming of chronic diseases.
Whether the same holds true for brain health is currently unknown, but there is intriguing experimental and epidemiological evidence suggesting a role for neuroendocrine stress in modifying associations with environmental contaminants. Male pups exposed prenatally to diesel exhaust and maternal stress exhibited microglial activation as well as cognitive impairments, effects not observed in female pups or following exposure to either diesel exhaust or stress alone [141]. Interestingly, serum corticosterone was higher at postnatal day 1 only in the male pups exposed prenatally to stress, suggesting a possible early role in priming microglial immune responses to the pollutant. In a recent population study, the association between PM2.5 and lower cognitive scores was found to be stronger among older adults living in higher stress neighborhoods [142]. Earlier work showed that neighborhood psychosocial stressors or individual perceived stress strengthened associations between tibia or blood lead levels and poorer performance on cognitive tests in older adults [143, 144], consistent with experimental findings of a modifying role for stress in neuroendocrine effects of lead [145 –147]. Co-exposure to air pollutants and noise, an important non-chemical stressor, was associated with greater impacts on cognitive function in adults aged 45–75 than would be expected by addition of independent effects, suggesting potential synergism [148]. Air pollutants have themselves been found to be associated with increased perceived stress [149], consistent with the notion that air pollution acts as a stressor and contributes to cumulative stress exposure. If prior or concurrent exposures to other stressors exacerbate the effects of pollutants on health outcomes such as cognitive impairment, impacts of pollutants may be greater in populations disproportionately exposed to stressors.
IMPLICATIONS AND KNOWLEDGE GAPS
Air pollution research has long been organ- or disease-specific, focused primarily on pulmonary and cardiometabolic diseases, and more recently on impacts on the brain including cognitive decline, dementia, and depression. Clearly, there is value in examining tissue- and disease-specific mechanisms; for example, understanding that nanoparticles may translocate to the brain via olfactory nerves [18] provides important insight into a potential route of exposure. Furthermore, it is clear that effects may differ according to dose and duration of exposure for different anatomical sites. In addition to such targeted studies, complementary approaches that consider effects of pollutants on systems that broadly impact the body are also needed to understand how the myriad health outcomes associated with exposure to air pollutants are connected, and to what extent they are driven by common initiating events. Despite distinct elements of neurological/mental health diseases, and considerable heterogeneity within a disease, a number of common underlying molecular and cellular mechanisms may increase susceptibility. These include dysregulated stress response systems [9], which are implicated also in a broad range of diseases that encompass those associated with air pollutants. It is unknown whether common initiating processes underlie the relationship between air pollutants and various diseases, although oxidative stress and inflammation have been identified as characteristic features of many disease states. Emerging evidence supports the notion that air pollutants act as chronic stressors, triggering stress response systems that, when chronically activated, contribute to a variety of disease states including those affecting the brain [8].
Assessing to what extent the health impacts of air pollutants are mediated through the stress axis may be helpful in improving our understanding of how pollutant exposure is associated with such a range of disease outcomes, and why adverse effects are seen in some individuals and not others. Because of their systemic nature, effects of chronic stressors will manifest in a variety of ways that depend not only on the nature of the stressor itself, but also on individual susceptibility. Exposure to stressors does not necessarily imply adverse consequences to the individual; rather, interindividual differences in susceptibility and resilience must be considered. There is substantial heterogeneity in stress reactivity in the human population [150]. Intrinsic factors such as age, sex, gender, stress reactivity, and existing disease, and extrinsic factors such as co-exposure to psychosocial and other stressors, may all potentially contribute to individual responses to pollutants. In rodent models, interstrain differences in stress axis function are associated with differences in lung injury and inflammatory response following exposure to ozone [151], suggesting that such variability may be relevant to susceptibility to pollutants. It will be important to assess to what extent interindividual differences are related to susceptibility to CNS impacts of air pollutants. Exposures during vulnerable windows of development are important determinants of future disease risk; indeed, stressors can produce epigenetic changes that influence future stress reactivity and morbidity, with some evidence that such effects may be transmitted across generations [152]. The timing, dose, pattern, and duration of exposure are likely to be important factors in determining effects, as seemingly opposing effects (e.g., pro- versus anti-inflammatory effects of glucocorticoids [47]) may be produced depending upon context. In the population setting, composite measures such as allostatic load indices may be useful in assessing potential chronic effects of multiple stressors that act through diverse biological mechanisms. In addition, interactions with established risk factors warrant investigation.
If stressor action by air pollutants is an important contributor to health impacts, characteristics that define the nature, magnitude, and duration of the stress response warrant investigation. Recent findings from a panel study show associations between specific particulate constituents, especially water-soluble inorganic ions, and stress hormone levels [153]. Bacterial lipopolysaccharide (LPS) is a potent stimulator of the HPA axis via its inflammatory effects [154], and is a common constituent of ambient particulate matter. There are high levels of airborne LPS in Mexico City, where exposure to pollution has been associated with a range of adverse impacts on the brain [17]. As effects of particulate matter and ozone on the stress axis were additive in experimental models [44], the combined effects of exposure to multiple pollutants should be considered. Both population and toxicological studies will be needed to investigate and identify factors driving stress responses.
CONCLUSION
Chronic activation and dysfunction of the stress response system is a characteristic of many disease processes, including those associated with air pollution. Observations of similar systemic responses to particulate and gaseous pollutants suggest that the endocrine stress response could be a common mechanism contributing to extrapulmonary effects of pollutant exposure, including impacts on the brain [8, 44]. Further research is warranted to investigate the relative importance of stress responses and HPA axis dysregulation in contributing to air pollutant-induced disease progression. An exciting implication of this work is that air pollution, like other stressors, could be a modifiable risk factor for neurodegenerative and psychiatric disorders. Efforts aimed at reducing stressor exposure at individual, neighborhood, and community levels could have the added benefit of decreasing vulnerability to the adverse health impacts of air pollutants. Future epidemiological and experimental studies should provide insight into such questions.
Footnotes
ACKNOWLEDGMENTS
The author is grateful for the contributions of collaborators, postdoctoral fellows, and students to the studies that formed the basis of this paper. Thanks to Drs. Dalibor Breznan, Guillaume Pelletier, and Jith Thomas for their helpful comments on the manuscript. Thanks also to the anonymous peer-reviewers, whose generous and constructive comments helped improve this paper. The research projects that led to this work were funded by Health Canada through the Clean Air Regulatory Agenda.