
Abstract
AIM:
Esophageal Squamous Cell Carcinoma (ESCC) is a histological subtype of esophageal cancer that begins in the squamous cells in the esophagus. In only 19% of the ESCC-diagnosed patients, a five-year survival rate has been seen. This necessitates the identification of high-confidence biomarkers for early diagnosis, prognosis, and potential therapeutic targets for the mitigation of ESCC.
METHOD:
We performed a meta-analysis of 10 mRNA datasets and identified consistently perturbed genes across the studies. Then, integrated with ESCC ATLAS to segregate ‘core’ genes to identify consequences of primary gene perturbation events leading to gene-gene interactions and dysregulated molecular signaling pathways. Further, by integrating with toxicogenomics data, inferences were drawn for gene interaction with environmental exposures, trace elements, chemical carcinogens, and drug chemicals. We also deduce the clinical outcomes of candidate genes based on survival analysis using the ESCC related dataset in The Cancer Genome Atlas.
RESULT:
We identified 237 known and 18 novel perturbed candidate genes. Desmoglein 1 (DSG1) is one such gene that we found significantly downregulated (Fold Change
CONCLUSION:
We identified novel perturbed genes in relation to ESCC and explored their interaction network. DSG1 is one such gene, its association with microbiota and a clinical presentation seen commonly with ESCC hints that it is a good candidate for early diagnostic marker. Besides, in this study we highlight candidate genes and their molecular connections to risk factors, biological pathways, drug chemicals, and the survival probability of ESCC patients.
Introduction
Esophageal Squamous cell carcinoma (ESCC) is an epidermoid carcinoma, most often found in the upper and middle part of the esophagus. This malignancy turns out deadly primarily due to late-stage diagnosis, metastasis, resistance to treatment, and relapse of cancer cells [1]. ESCC is the most common form of the two types of esophageal cancer with high prevalence in East Asian, Eastern European, and African populations, whereas less prevalent in Central European and Hispanic populations. Importantly, two geographic belts, the Asian esophageal cancer belt across central Asia from the Caspian Sea to northern China, and a belt on the eastern coast of Africa, from Ethiopia to South Africa, were found to have the highest risk for ESCC [2].
Lifestyle factors including smoking, chewing tobacco or gutka (a preparation made of crushed areca nut, tobacco, catechu, paraffin wax, slaked lime, and sweet or savory flavoring), consumption of heavy alcohol, and hot drinks are considered to be the critical risk factors for ESCC [3, 4, 5]. Further, low intake of fresh fruits and vegetables, pickled vegetables, exposure to polycyclic aromatic hydrocarbons, and betel quid chewing with or without tobacco [6] also have been reported to be risk factors for ESCC. Notably, many of these risk factors are geographical region specific [7]. For example, consumption of salted meat and its interaction with alcohol intake and smoking was found to be a risk factor for ESCC in Uganda [8], trace element imbalance in the soils in South Africa and West Asia [7], frequent drinking of hot Arabic coffee in Al-Qaseem region of the Saudi Arabia [9], consumption of hot green tea in East Asia and betel quid/gutka chewing in regions of South and Southeast Asia [10]. In addition, dietary zinc deficiency is known to increase the risk of ESCC [11]. The serum levels of Selenium and Zinc correlate with incidence of gastroesophageal cancers in West Asia [12].
In our previous effort, we developed ESCC ATLAS, a manually curated database, for integrating genetic, epigenetic, transcriptomic and proteomic knowledge disposed of in published literature [13]. It comprised 3,475 genes with the evidence of altered transcription (2,600), altered translation (560), and associated with molecular events such as copy number variation/structural variations (233), SNPs (102), altered DNA methylation (82), Histone modifications (16) and miRNA-based regulation (261) in ESCC etiology. Inarguably, biomarkers are valuable in predicting the outcome and guiding treatments of diseases. However, only a few biomarkers such as epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF), epidermal growth factor receptor 3 (HER3), programmed death receptor 1 or PD-L1 programmed death-ligand 1 (PD-1/PD-L1) serve as a therapeutic target in ESCC treatment [14] despite severe side effects from drugs in patients. The standard chemotherapeutic drugs used in treatment for ESCC are Cisplatin, Docetaxel, Mitomycin, 5-Fluorouracil, and vinorelbine or their combinations. The combination of docetaxel-cisplatin has shown a higher pathological complete response rate than the fluorouracil-cisplatin combination [15].
Often the case in the setting of genome-scale studies, genes are filtered based on their lowest p-values to select the top list of genes. As a result, only a few genes are selected for the validation and functional characterization phase of the study. If these genes replicate in an independent cohort, they are considered “perturbed genes” and are reported in the main manuscript/research. Sadly, the rest of the potentially important genes falling right below the cream layer never become the focus of the follow-up studies. An important point that should be noted is that the number of genes that fall in the top hits list usually depends on the sample size of the study. Hence, when the genes that stand just below a p-value threshold are not considered, there is a high chance that a potentially high-value biomarker gene is not considered for further investigation. Hence, hypotheses inferred and validated based solely on the results of a single study could sometimes be incomplete or misleading. Typically, the results vary between studies. Hence, instead of a single study, drawing conclusions based on combined data across studies would offer more reliable results.
In this contribution, we performed a meta-analysis using the data collected from multiple published studies. Here we present a list of differentially perturbed genes across multiple datasets in context to the ESCC, and explore their gene-to-gene interaction network, toxicogenomic targets in ESCC etiology and treatment, and report their association with specific biological pathways and GO terms.
Materials and methods
Source and collection of datasets
Studies related to mRNA expression of human ESCC tissues and cell lines were searched using the following keywords – “ESCC” or “Esophageal Squamous Cell Carcinoma” in three different public gene expression databases – 1) Gene Expression Omnibus (NCBI-GEO) [16], 2) EBI ArrayExpress [17] and 3) All of gene expression (AOE) [18] with certain criteria; which included – 1) selection of studies of human origin involving normal-tumor/cell lines samples were selected 2) the drug-treated and gene knockdown studies were excluded, 3) only the protein-coding mRNA studies were included, whereas the and non-coding RNA/miRNA studies were excluded 4) studies with at least two normal and tumor samples were selected. Based on this, a total of 16 datasets, comprising 782 (of which 298 normal, 484 tumor) samples altogether, were selected and preprocessed for quality control in our current study. However, 10 of the 16 datasets were further selected based on the quality control measures for downstream analysis.
Quality control measures and tools for analysis
The quality check of the Microarray datasets was performed using arrayQualityMetrics (v3.48.0) [19], a Bioconductor package that provides a detailed array quality metrics report with diagnostic plots using six different outlier detection methods. The arrayQualityMetrics also flags the potential outlier samples; considering the outlier flags and manual inspection the samples were removed from the array (Supplementary Table S1). The dataset itself was discarded if a potential mislabeling sample mislabeling was identified or if it contained an unbalanced number of control and tumor samples. In the case of the RNAseq dataset (GSE32424), we referred to the quality check performed in the original study and directly used the normalized read counts available from GEO [20].
As our collection of datasets was produced from different platforms, the platform-specific methods for processing the dataset were implemented. Hence, the datasets produced using the Affymetrix array platform were analyzed using the Affy (v1.74.0) [21] Bioconductor package in R. Similarly, the data generated from the Agilent, Human Whole Genome Onearray (HOA), and CodeLink array platforms, were processed using the LIMMA (v3.52.2) [22, 23] Bioconductor package. Each dataset was subjected to background correction, followed by the platform-specific normalization methods. The selection of the normalization method was based on the data distributions in Table 2.
Data enhancement and annotation
All the raw datasets were subjected to platform specific background correction and normalization methods. For this, all the available normalization methods were compared and only the optimal normalization method that provided a normal distribution (or approximate normal distribution) of data was selected. The resultant data was used for further analysis. Multiple probes in each dataset were collapsed to their gene source either by taking mean or median value of probeset intensities. The choice of mean or median depended upon the data distribution (observation made using density plots). Gene mapping to its probe IDs across studies was done by matching the official gene symbols using either platform specific design files or Bioconductor annotation packages including AnnotationDBI (v1.58.0) [24].
Differential gene expression of individual datasets and Meta-analysis
To discover differentially expressed genes, contrast matrices with tumor
To identify genes that are either known to be associated with esophageal squamous cell carcinoma (ESCC) or are potentially novel, we compared differentially expressed genes to those that have previously been reported in ESCC ATLAS. Additionally, we evaluated the expression differences of these candidate novel genes across different tumor cell differentiability statuses (Poor, Moderate, and Well) and normal tissue using the GSE75241 dataset. The comparison was made using a Wilcoxon rank sum test, where the differentiability statuses range from being cancerous (Poor) to normal (Well).
Gene to gene network analysis
To identify if our candidate genes were associated to the ESCC related molecular events, such as – 1) structural variations, 2) SNPs, 3) altered DNA methylation, 4) Histone modifications or 5) miRNA targets, they were intersected against the genes curated in the ESCC ATLAS [13], as well as a list of additional mutated genes reported in ESCC studies that were not yet included in ESCC ATLAS [28, 29, 30, 31]. The genes mapping to these molecular events were considered as the ‘core’ genes, whereas the remaining as ‘peripheral’ genes.
The gene-to-gene interactions between ‘core’ and ‘peripheral’ genes were investigated using a gene-to-gene network generated by querying the STRING database using the Cytoscape (v3.7.2) StringApp (v1.6.0) (The minimum interaction confidence score was set to 0.6) [32]. In the network each node represented the candidate genes. The nodes were colored with a color scale representing the average Fold Change (FC) expression across all the studies that were obtained from the meta-analysis using the Omics Visualizer (v1.3.0). We also explored the gene-disease networks for the selected genes using the DisGeNET Cytoscape App (v7.3.0) [33]. The gene-disease network is bipartite graph with two types of vertices (genes and diseases) and the edges connecting the vertices of different types (e.g., a gene with a disease). In this graph, two vertices can be connected by more than one edge, representing the multiple pieces of evidence reporting the gene-disease and variant-disease associations such as different database sources or DisGeNET association type [33].
Functional enrichment analysis
To gain insights into the biological functions and molecular networks that may have been altered in ESCC etiology for the top 255 differential expressed genes, they were evaluated for the overrepresented GO terms using gseGO, KEGG pathways using gseKEGG, Reactome pathways using gsePathway and WikiPathways using gseWP functions from the ClusterProfiler (v4.4.4) [34] package. These functions compute the overrepresentation based on the hypergeometric distribution test; the p-values were adjusted for the multiple testing in each case using the Benjamini-Hochberg (BH) procedure. Finally, the candidate gene set was selected considering the threshold on the adjusted
Toxicogenomics analysis
The interactions of the candidate genes with the environmental exposures (such as chemical carcinogens, tobacco smoke, trace elements deficiency) and drugs that are inferred in context to ESCC, were explored using the bioconductor package CTDquerier (v2.3.1) [35] and represented in the form of a network, using the information from the Comparative Toxicogenomic Database [36]. For the network representation we used the ggnet package (v0.1.0) in R.
Characteristics of datasets used in the meta-analysis
Characteristics of datasets used in the meta-analysis
To reveal contribution of candidate genes on patient survival, we performed overall survival prediction in an independent ESCC RNASeq dataset available at The Cancer Genome Atlas (TCGA). We used TCGAbiolinks (v2.20.1) in R [37] to access 90 ESCC tumor and 11 normal samples [38] of the total 559 samples corresponding to EC. Further, we used the survival package (v 0.4.9) and survminer package (v3.4.0) in R for survival analysis and for generating Kaplan–Meier survival plots respectively. For the genes that showed significant association with survival of ESCC patients, we performed Cox proportional hazard analyses using the dysregulated and intact candidate genes, and compared the distribution of log2CPM values for these genes in the tumor and normal samples using
Results
Characteristics of datasets
We collected the data available from a total of 16 studies that were selected with our exhaustive literature survey and selection criteria (see Methods). These studies differed from each other in terms of samples, experimental setup and the data generation platforms. 13 studies used the normal-tumor tissue samples, whereas the other 3 used the ESCC cell lines. Of the 16 datasets, 11 were generated from the Affymetrix platform, and the remaining 5 datasets were from Agilent, Phalanx Human OneArray, Codelink, and Illumina Genome Analyzer platforms (1 each. A detailed Information on the datasets with biological materials, sample size, microarray/sequencing platforms used in each study is provided in Table 1.
QQ-plot showing observed versus theoretical quantiles of expression in all the 10 ESCC ‘normal vs tumor’ datasets.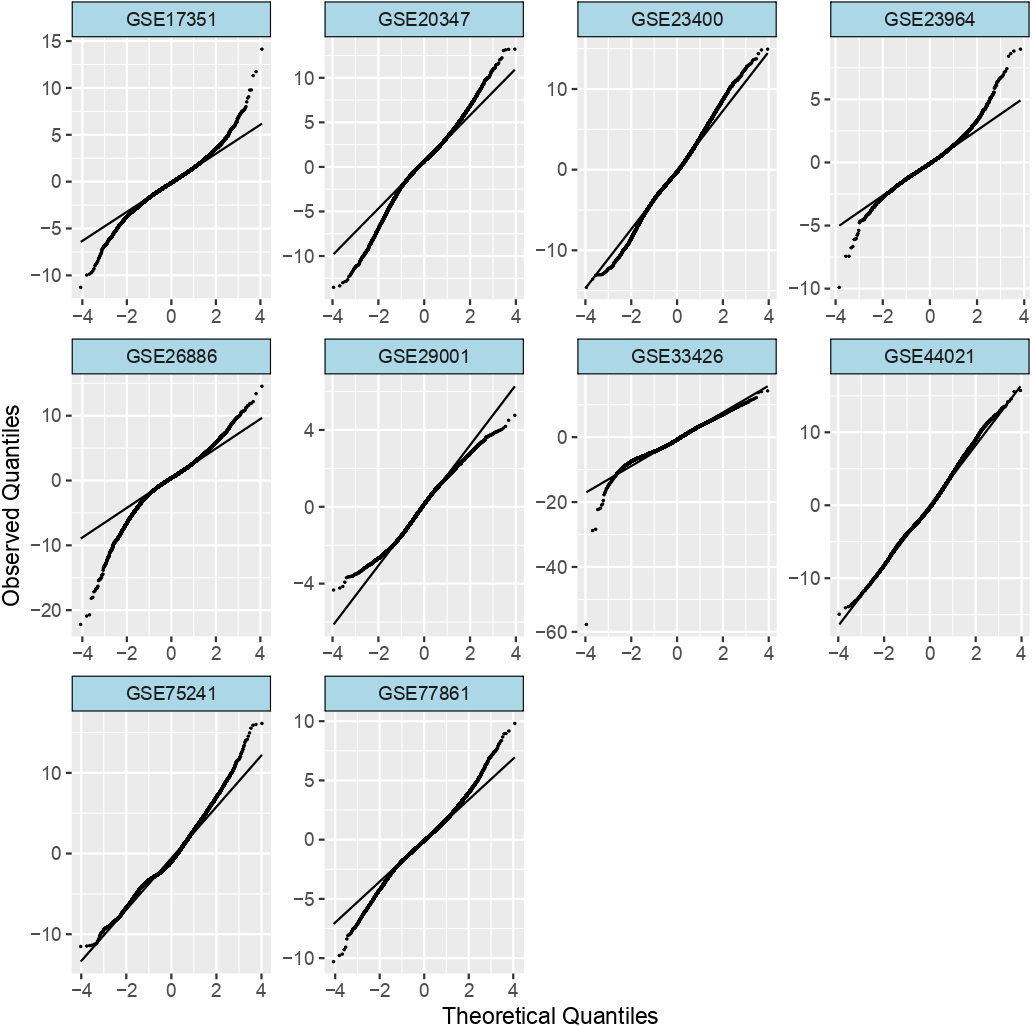
For any meta-analysis it is crucial to assess the quality of the dataset being used, because the inclusion of an outlying dataset could result in inefficient or wrong biological conclusions [39]. Hence, we performed the quality check for each of our collected datasets (see Methods), such that there were no outlier samples in the dataset and the data itself were not unbalanced. Considering the stringent QC measures and the number of common gene set across the arrays, we disregarded 6 datasets for further analysis, these included – GSE63941, GSE9982, GSE45168, GSE70409, GSE22954 and GSE32424 (Supplementary Table S1), among these we found possible mislabeling of the samples in GSE9982, GSE45168 (Supplementary Figure S1). The details of the number of samples being excluded in each dataset and QC status of the datasets are provided in Table 2.
Illustrates study specific details of the analysis method used and number of genes identified from original studies and this study
Note #, all the normal samples failed in QC parameters, $ mislabeling was suspected in sample status, @datasets were removed for comprising less number of genes in those datasets, N refers to ‘normal’ samples and T refers to ‘tumor’ samples, * Test for symmetry between DE gene distribution between original and this study using McNemar–Bowker chi-square test.
Heatmap showing differential expression of the most consistently perturbed 255 genes across 10 datasets.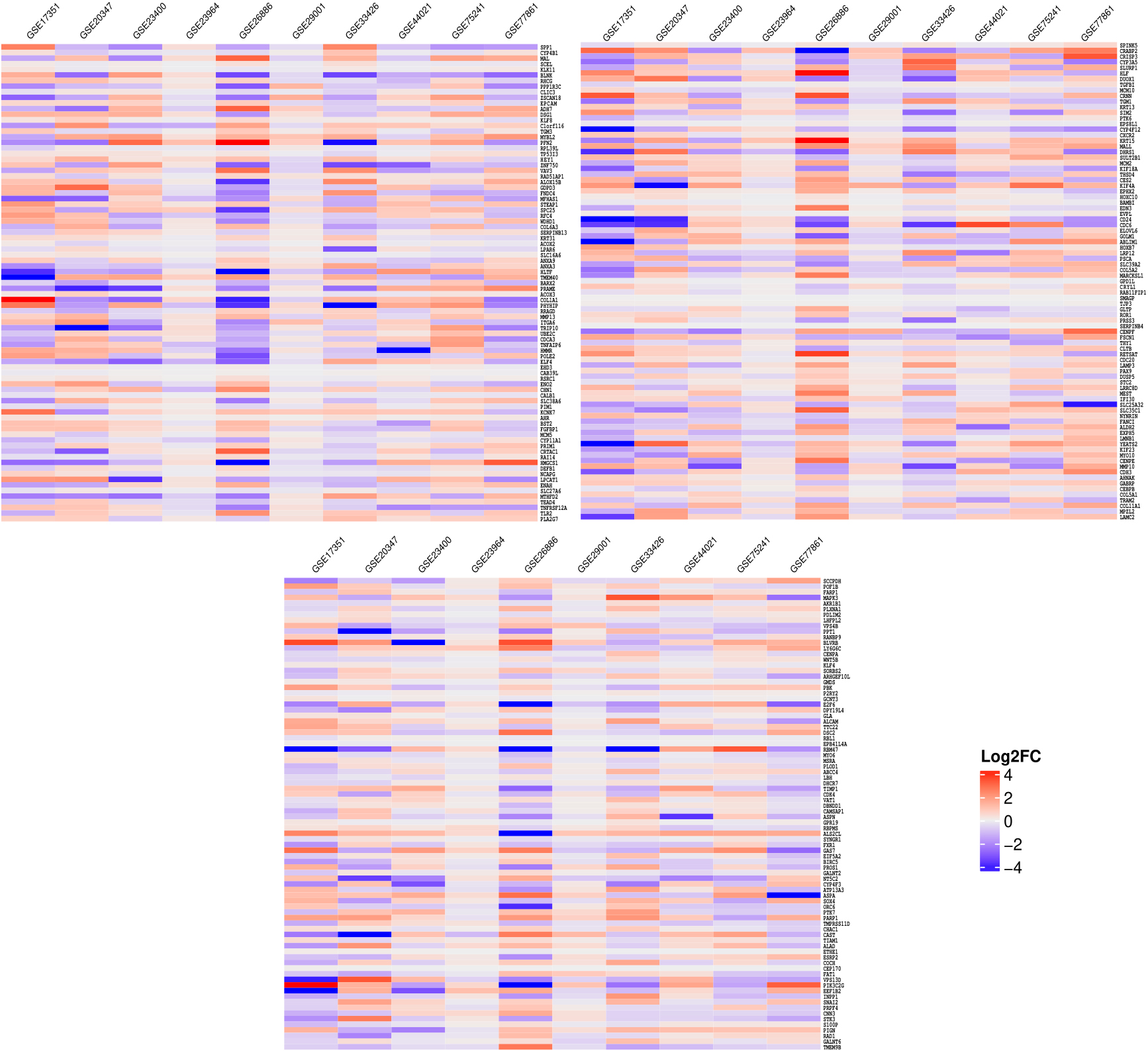
We identified the differentially expressed genes in each dataset based on the linear modelling of tumor
List of genes consistently perturbed with
1.5 and
1.5 summary fold change
List of genes consistently perturbed with
Showing A) Forest plot showing the expression of the DSG1 consistently downregulated in all the studies with at least 6 of the studies showing significant downregulation (p-value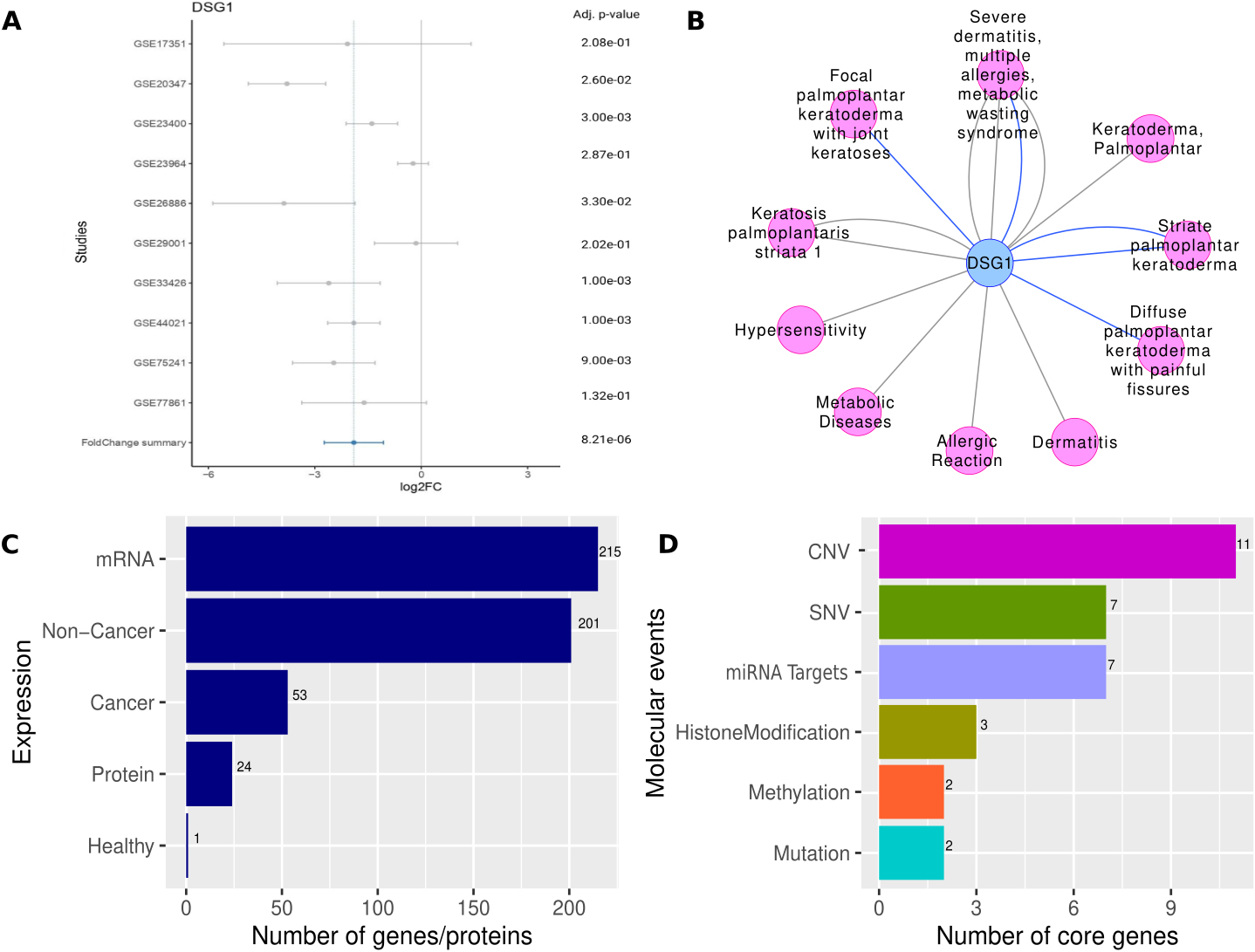
To identify if our candidate genes set were previously known in context to ESCC or were associated to the ESCC related molecular events, we queried them against the ESCC ATLAS [13] and other additional studies that were not included in the ESCC ATLAS. We found, 237 of our 255 candidate genes were indeed known and listed in ESCC ATLAS. More importantly, we also found 18 perturbed genes (DSG1, RETSAT, KCNK7, AKR1B1, LY6G6C, GLA, TTC22, DBNDD1, GPR19, PROS1, GALNT2, ESRP2, COCH, VPS13D, PRPF4, PIGN, RAD1, and GALNT6) that were not known or reported in context to ESCC, and hence they are important genes to be further investigated.
Showing interaction network of core genes with peripheral genes. The edges connecting the core gene and peripheral genes are highlighted; the color of the nodes represent the average log2FC of the genes across studies. The nodes corresponding to the core genes were highlighted with distinct colored squares, where the colors of the squares corresponded to specific molecular events.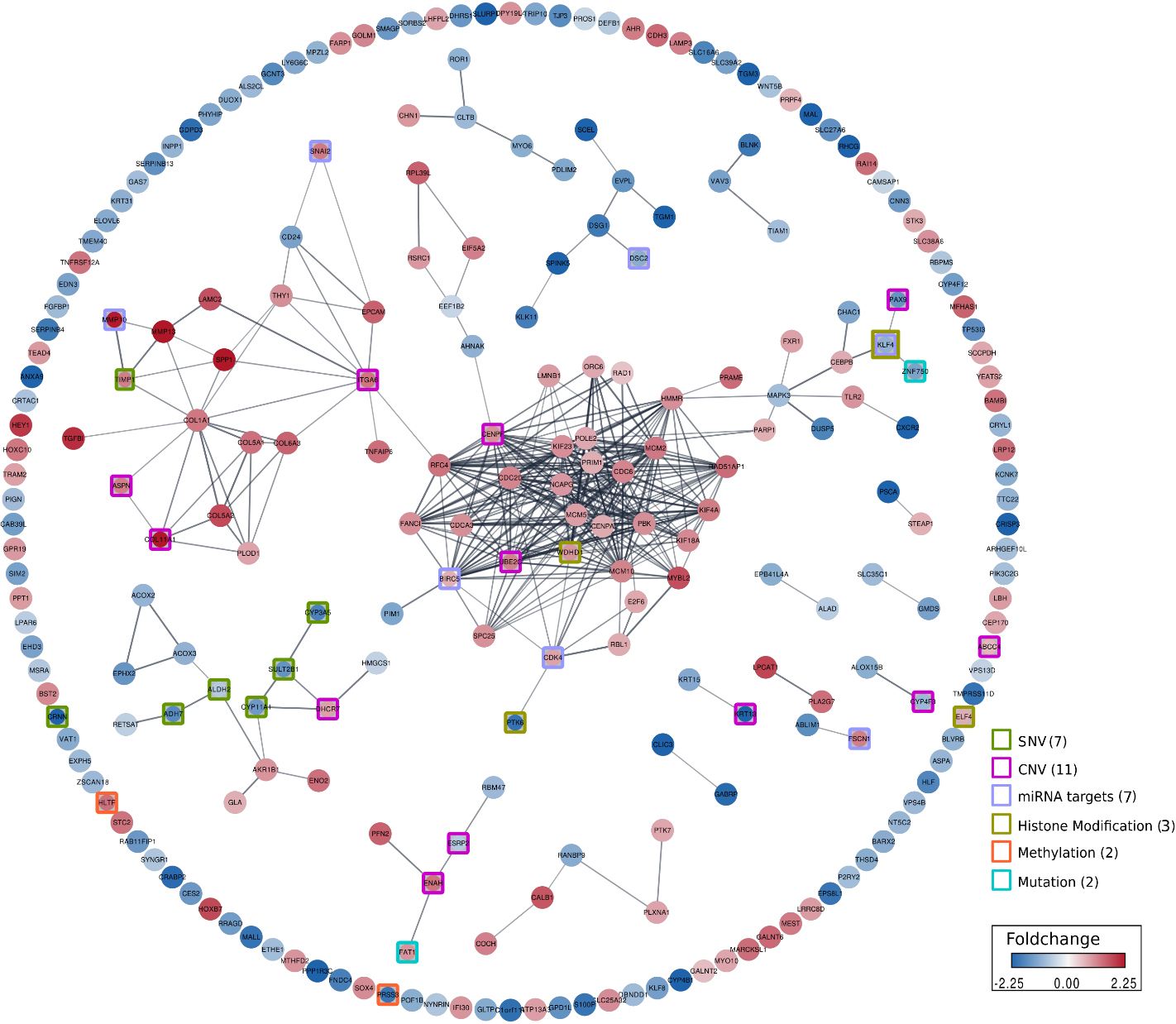
We assessed the expression differences of these novel perturbed genes between tumor cell differentiation status and normal tissue using the data available from GSE75241. The results revealed significant differences in the expression of AKR1B1, DBNDD1, DSG1, ESRP2, GALNT2, GALNT6, LY6G6C, PIGN, RETSAT, and TTC22 genes (Supplementary Figure 3). The observation that these genes exhibit significant differences in their expression levels between tumor cell differentiation status (Moderate, Poor, and Well) and normal tissue suggests that they play important role in tumor cell differentiation and morphogenesis as well as embryonic development through their ability to influence intercellular signal transduction in ESCC.
Illustrating functional gene enrichment analysis of overrepresentation terms from A) GO enrichment B) KEGG C) Wikipathways enrichment D) Reactome.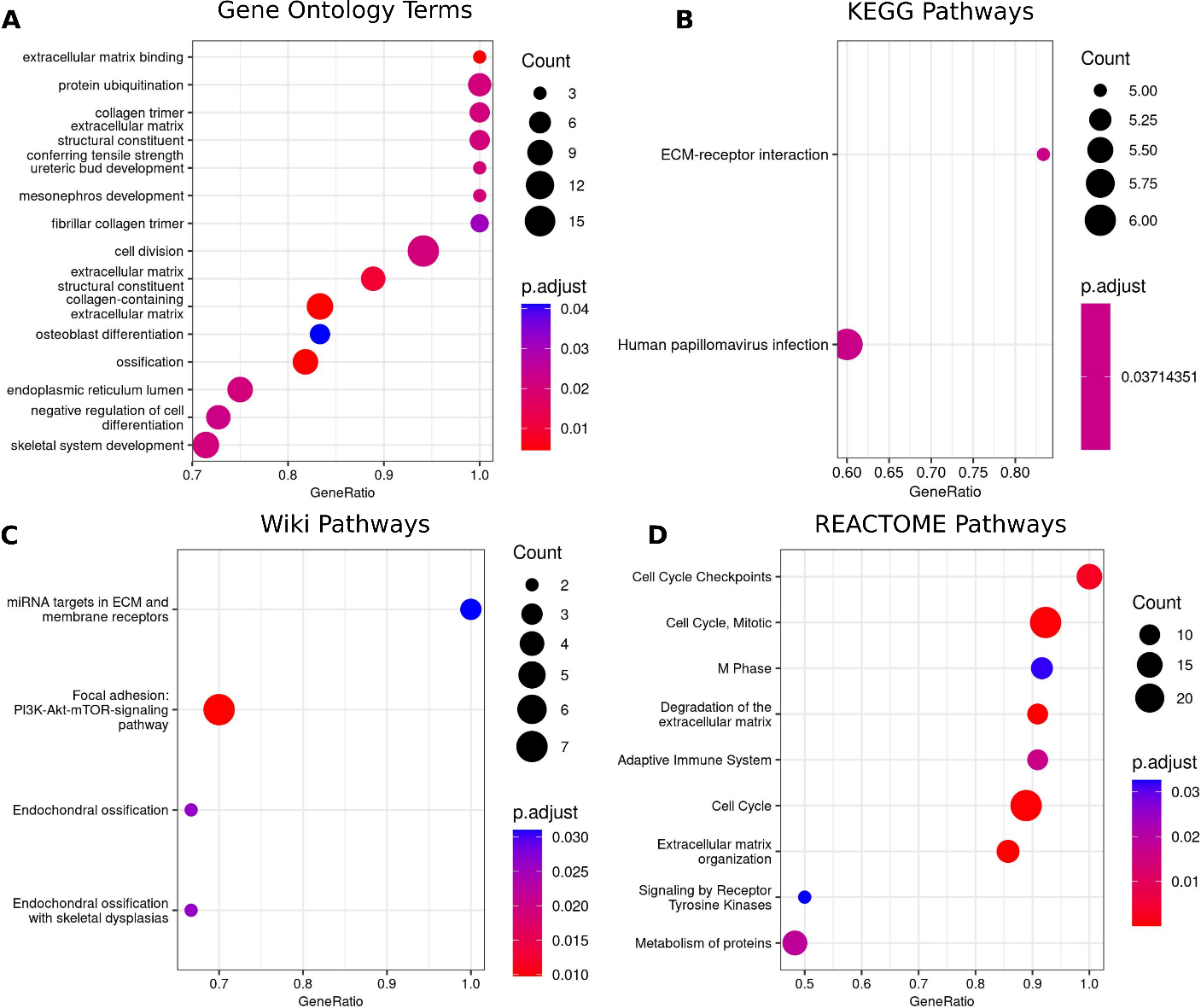
We also referred to the Cancer Dependency Map (DepMap) [40] portal which includes the effect size information for a given gene that would represent how much the loss of that gene affects the viability of the cancer cell line being studied. Of all the 18 genes only Desmoglein-1 (DSG1) gene knockout in DepMap was related to the ESCC cell line that was compared against the 24 different cell lineage-specific gene sets to calculate the gene dependency scores. We found the Gene Effect (Chronos) for DSG1 in ESCC was significantly different from the other lineage-specific gene sets and less than zero, indicating that DSG1 is essential in ESCC (Supplementary Figure 4).
Among these, Desmoglein-1 (DSG1) showed significant downregulation (FC
By intersecting our candidate gene lists with the NSG 7.0, which catalogues cancer-healthy driver genes, we observed 53 canonical cancer genes, 201 non-cancer genes and one healthy driver genes in context to ESCC. This suggested that cancer genes were enriched (
Lists of biological terms with summary statistics of functional enrichment analysis using ClusterProfiler
Further, we segregated our candidate genes (see Methods) set as core (genes that are known to harbor genetic variants or undergo epigenetic regulation in ESCC ATLAS) and peripheral genes (other perturbed genes). In total we found 31 core genes, of which 7 were previously known to harbor SNPs in context to ESCC (namely, CRNN, CYP3A5, ADH7, SULT2B1, ALDH2, CYP11A1, MMP7), whereas other 11 genes were known to be associated with CNVs (namely, KRT13, ITGA6, UBE2C, CENPF, PAX9, ENAH, COL11A1, ABCC4, DHCR7, ASPN, CYP4F3) in relation to ESCC. Additionally, we identified 11 genes with either epigenetic modifications such as DNA methylation (namely, PRSS3, HLTF), Histone Modification (include, PTK6, WDHD1, KLF4), or targets of miRNAs (FSCN1, MMP10, KLF4, DSC2, CDK4, SNAI2, BIRC5). We also found 2 genes, FAT1 and ZNF750 that were associated with mutations in context to ESCC. The distribution of core genes with respect to molecular events were shown in Fig. 3D. Details of core genes concerning molecular events in the top 255 genes with curated information from the original studies are presented in Supplementary Dataset S1. We could map 215 mRNA genes and protein expression information for 24 genes in ESCC ATLAS (Fig. 3C).
Next, we explored the gene-to-gene interaction network, where the “core” and “peripheral” genes were interconnected based on the available evidence for functional associations or physical interactions between protein-protein pairs imported by the StringApp from the STRING database into Cytoscape. We found, of 31 total core genes, 27 interact with at least one other peripheral or core gene that is differentially regulated (Fig. 4). Whereas, 132 differentially regulated genes including 7 core genes did not have enough evidence for the interaction with other genes. Within our network of interacting genes, we found a sub-network of collagen type 1, 5, 6 and 11 genes that are all upregulated in ESCC. Collagen type XI alpha 1 chain (COL11A1) which is one of the core genes, is previously known for promoting ESCC proliferation, and for the target of miRNA-based regulation [43], and also to be associated with CNV in context to ESCC [44]. Collagen gene family are also known to be upregulated for the synthesis and assembly of collagens driven by tumor-associated macrophages (TAMs) which are in turn involved in the extracellular matrix (ECM) remodeling proteases in the primary tumor environment [45]. Another interesting gene in our network is the transcriptional regulator Krüppel-like factor 4 (KLF4) that is previously known to be downregulated in ESCC [46] and is also known to interact with miRNAs-based regulation [49]. We found KLF4 interacts with ZNF750 which is also a core gene, and is a known transcriptional regulator of epidermal differentiation. ZNF750 has been previously identified as a tumor suppressor in ESCC [47]. Several reports have shown that ZNF750 positively regulates KLF4 expression [48, 49]. Hence it is not surprising to see these two genes are downregulated together in the context to ESCC [49, 50].
Functional enrichment of genes
Gene perturbation driven tumultuous molecular responses discovered from functional enrichment analysis have shown activation of crucial biological processes and pathways in ESCC etiology. The enrichment analysis using the GO annotations showed 15 terms to be significantly (adjusted
Toxicogenomic database inferences for the candidate gene set showing genes interaction with risk factors of ESCC and drug chemicals used in treatment of ESCC. Strong color gradient for the genes indicate high inference score whereas light color indicate low inference score. 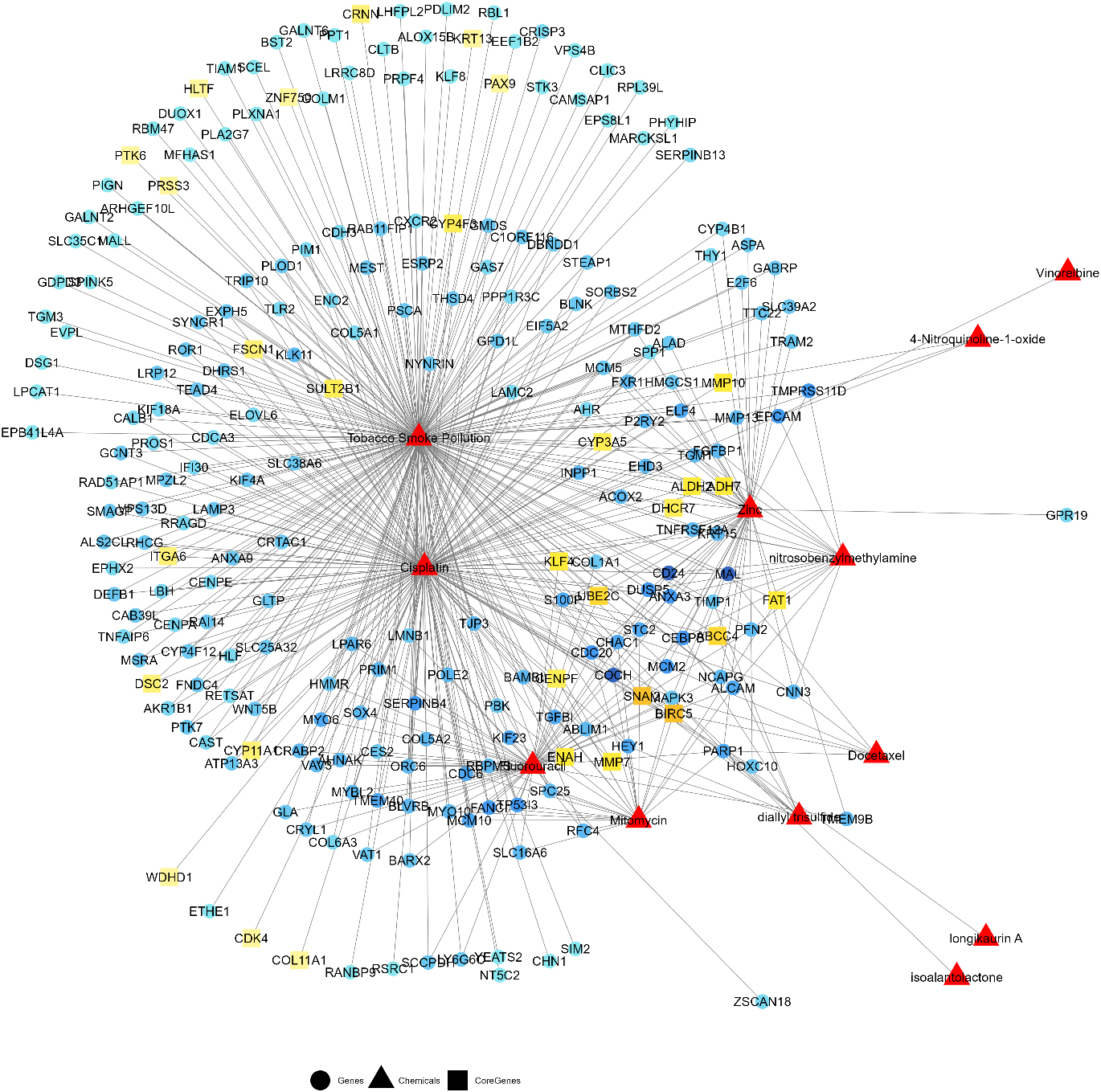
Evidently, literature reports support implication of these terms in cancer etiology, more particularly in ESCC. For example – i) Extracellular matrix remodeling: Tumor-induced ECM alterations are a hallmark of malignancy. It increases tissue stiffness and desmoplasia around the tumor [45]. ii) Ossification: the abnormal development of bone tissue within periarticular soft tissue is reported in some cancers [51] albeit not well reported in ESCC. iii) Protein ubiquitination: ubiquitination is the second most common post translational modification (PTM) for proteins and aberrant ubiquitination may lead to cancer development and progression [52]. iv) Collagen trimer: Collagen is a key structural component of ECM and its breakdown by matrix metalloproteinases (MMPs) and other proteases promote angiogenesis and tumor invasion [53]. v) Cell cycle, cell division and negative regulation of cell differentiation: alteration in the cell-cycle and apoptotic machineries allow cancer cells to escape the normal control of cell proliferation and cell death. vi) Human papillomavirus infection: HPV infection is a known risk factor for ESCC [54]. vii) Adaptive Immune System: Tumor-associated antigens are triggers of the immune response. They activate the T cell response, an important line of defense against tumorigenesis [55]. viii) Signalling by Receptor Tyrosine Kinases (RTKs): RTKs are a family of integral cell surface membrane receptors. They are essential to mediate cell-to-cell communication and play key roles in cellular growth, differentiation, metabolism and motility [56]. ix) Focal adhesion: PI3K-Akt-mTOR-signaling pathway: This pathway plays a crucial role in the regulation of cell growth, differentiation, migration, metabolism and proliferation. The PI3K/Akt/mTOR pathway is known to be activated in the development and progression of ESCC [57].
The Kaplan Meier plot showing differences in overall survival between dysregulated vs intact expression of genes with survival curves. Genes including A) MCM10, B) CEP170 shown survival benefit whereas C) STC2 and D) DBNDD1 shown survival risk in 90 ESCC patients using their RNAseq based gene expression and clinical data from TCGA.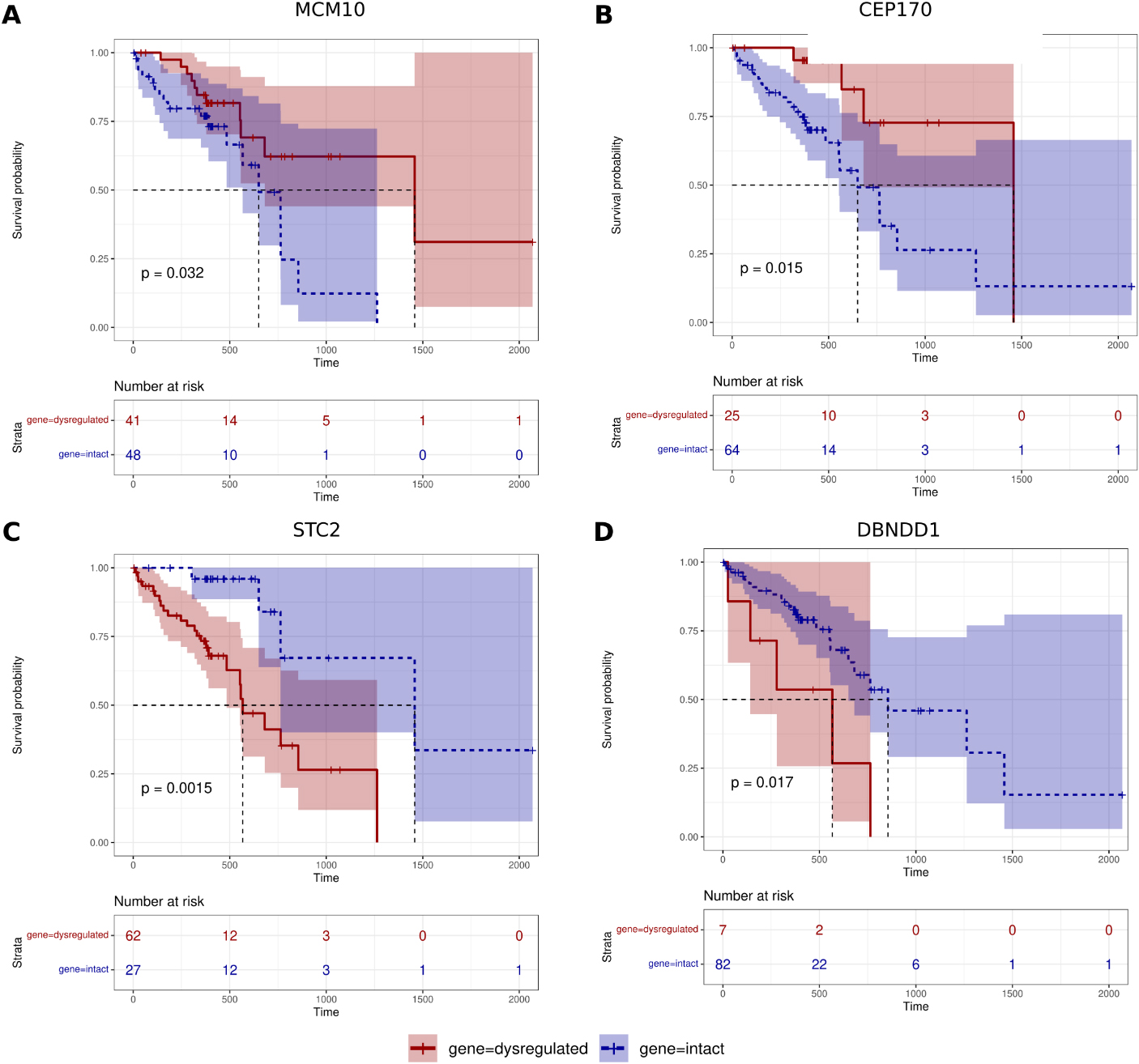
In order to investigate the chemical-gene interactions for our candidate gene set we referred to the Comparative Toxicogenomics Database (CTD) [58, 59] that catalogs the manually curated information about chemical – gene/protein interactions, chemical – disease and gene – disease relationships. We could map 240 candidate genes in the CTD. We found these genes were linked to various risk factors, chemical carcinogens and drugs in relation to ESCC, which includes – 223 genes associated with Tobacco Smoke Pollution, 18 genes with nitroso benzylmethylamine, 3 genes with 4-Nitroquinoline-1-oxide, 49 genes with trace element- Zinc, 168 genes with Cisplatin, 54 with Fluorouracil, 25 with Mitomycin, 6 with Docetaxel, 5 with diallyl trisulfide, 1 with each Vinorelbine, isoalantolactone, and longikaurin A) of therapeutic values in ESCC treatment. Among 240 gene relationships with chemicals, 4 genes (such as KRT13, ALDH2, BLVRB, FAT1) had ‘Direct’ evidence to chemical exposure. Of them, 3 genes (such as KRT13, ALDH2, BLVRB, and FAT1) were ‘core’ genes (see Supplementary Table S4). A network of interaction between differentially expressed genes and chemical exposures is illustrated in Fig. 6. An exercise of mapping core genes onto the network revealed the interaction of 26 core genes (CYP3A5, CRNN, ADH7, KRT13, PTK6, SULT2B1, ZNF750, HLTF, ITGA6, PRSS3, UBE2C, CENPF, FSCN1, PAX9, ALDH2, CYP11A1, MMP10, ENAH, KLF4, DSC2, DHCR7, BIRC5, CYP4F3, FAT1, SNAI2, MMP7) with Tobacco Smoke Pollution, 6 core genes (CYP3A5, UBE2C, ALDH2, KLF4, BIRC5, SNAI2) with Zinc, 4 core genes (ADH7, ALDH2, DHCR7, FAT1) with nitrosobenzylmethylamine and 1 core gene (MMP10) with 4-Nitroquinoline-1-oxide. Further, interaction of 21 core genes (CYP3A5, ADH7, SULT2B1, WDHD1, ITGA6, UBE2C, CENPF, FSCN1, ALDH2, CYP11A1, MMP10, ENAH, COL11A1, KLF4, DSC2, DHCR7, CDK4, BIRC5, CYP4F3, SNAI2, MMP7) with cisplatin, 5 (UBE2C, ENAH, KLF4, BIRC5, SNAI2) with Fluorouracil, 3 (BIRC5, SNAI2, MMP7) with diallyl trisulfide, 3 (CENPF, BIRC5, FAT1) with Mitomycin and 1 (BIRC5) with Docetaxel.
Identification of candidate genes associated with prognosis in ESCC
By using gene expression data of 90 ESCC patients from The Cancer Genome Atlas (TCGA) and a Cox proportional hazard regression analysis, we predicted the clinical outcomes of candidate genes in ESCC patients. In total, 4 genes – MCM10, CEP170, STC2 and DBNDD1, were significantly correlated with overall survival. We found upregulation of MCM10, and CEP170 have beneficial effect with Hazard Ratio (HR)
Cox proportional Hazard model for prognosis of ESCC in 90 patient’s data accessed from TCGA
Cox proportional Hazard model for prognosis of ESCC in 90 patient’s data accessed from TCGA
Here we presented a Meta-analysis of 10 globally published and unpublished mRNA expression of ESCC tumor-normal tissue datasets, and report 255 differentially regulated genes, of which we classify 31 as core genes are associated with the previously known molecular events in relation to ESCC. We explored the Gene-Gene interactions of the 255 candidate genes including core genes that are possibly involved in the perturbation of the interconnected genes to bring about phenotypic changes. We believe our meta-analysis and the gene interaction network presented here shed light on the new set of genes that have not been investigated in the context of ESCC so far. We report 18 such perturbed genes in ESCC etiology, of which DSG1 showed 1.9 fold downregulation. Functionally, desmogleins are cadherin-like transmembrane glycoproteins, along with desmocollins they form a major component of the intercellular junction – desmosome. Desmosomes are essential for epithelial differentiation and the loss of intercellular adhesion that facilitates tumor cell invasion and metastasis. The encoded protein has been identified as a target of auto-antibodies in the autoimmune skin blistering disease pemphigus foliaceus, a clinical presentation seen with esophageal cancer [60, 61]. Downregulation of this gene has also been observed with eosinophilic esophagitis (EE) (a chronic inflammation within the esophageal mucosa) and the silencing of DSG1 resulting into the weakening of esophageal epithelial integrity, which could result in the cell separation and impaired barrier function [62]. EE may predispose patients to malignant transformation. However, there are only a few case reports on co-occurrence of EE and ESCC [63]. In line with our observation in ESCC, DSG1 is also downregulated in anal carcinoma of squamous cells [64]. Additionally, DSG1 is also reported to be a receptor for Staphylococcus aureus cell wall anchored Serine-Aspartate repeat containing protein D (SdrD). The evidence presented in a study [65] suggests a strong association between squamous cell carcinoma and presence of S. aureus [65] , a Gram-positive spherically shaped bacterium frequently found in the upper respiratory tract and on the skin. In recent studies a crucial link between S. aureus and smoking has also has shown that, normally harmless S. aureus in humans while adopting to smoking induced oxidative stress, becomes more virulent [66] and infects persistently [67]. When all these crucial links are put together, DSG1 could be an important early diagnostic marker which was plausibly missed by the ESCC research studies so far. Here we also point at two other differentially regulated genes, TMPRSS11D and PRSS3 (belonging to a family of serine proteases) that are known for regulating the interaction between microbes and the immune system. In human hosts, TMPRSS11D and PRSS3 are known to mediate proteolytic activation and replication of viruses respectively [68, 69]. Further, while identifying plausible bacterial role (S. aureus), we identified links to plausible HPV infection in ESCC etiology, where the following set of genes SPP1, HEY1, LAMC2, COL6A3, ITGA6, COL1A1 were involved in the viral infection. In support of the molecular connections seen, a recent study of HPV infections in the oral cavity of mice progressing to the cancer stage in conjunction with environmental pollutants and tobacco smoke has been identified in oral squamous cell carcinoma [70].
Interestingly, many of the biological processes or pathways identified from the functional enrichment analysis are ‘bittersweet’ in function, i.e, they could possess a dual regulatory role in cancer. For example, ECM (which is consistently overrepresented in our candidate genes) components possess both tumor-suppressing and tumor-promoting properties [45]. This stands true also for the “protein ubiquitination”, since ubiquitination is a reversible process, it can be utilized in cell reprogramming [52]. We also point at a set of the collagen related genes (specifically COL11A1, which is upregulated with fold change 2.23) that could be a potential target for ESCC treatment [53]. We see that our candidate genes set also overrepresents the RTKs pathways. The genes involved in this pathway are potential pharmacological targets for the treatment of many malignancies associated with oncogenic activation of RTKs. Also, many small molecule RTK inhibitors are clinically approved for treatment of several cancers [56]. We have shown the PI3K-Akt-mTOR-signaling pathway genes are enriched in our candidate gene set, and the inhibitors (including Oridonin-as Akt inhibitor, Rapamycin-as mTORC1 inhibitor and BEZ235- as an ATP-competitive dual pan-PI3K and mTORC1/mTORC2 inhibitor) targeting PI3K/Akt/mTOR pathway are shown to play an important role in ESCC [57]. Therefore, the candidate genes we present here are important to be further investigated and could possess potential value in guiding the treatment for ESCC.
By using the CTD, here we also unraveled the interaction of our candidate gene set with several cancer-causing environmental risk factors, chemicals and drug targets. One of the interesting interactions include the GPR19 (novel perturbed gene in relation to ESCC) with trace element- Zinc. Prolonged Zinc deficiency is known to cause inflammation in esophageal mucosa. It is previously shown that the treatment with low doses of the environmental carcinogen, N-nitrosomethylbenzylamine (NMBA) in zinc-deficient rats, the incidence of ESCC increased to 66%, whereas, in case of zinc-sufficient rats, the low doses of NMBA showed no cancers [11]. Inferences from CTD showed 8 other differentially regulated genes - CD24, ANXA3, DUSP5, TNFRSF12A, ALDH2, CEBPB, TMPRSS11D, and CXCL10 also interact with zinc as well as nitroso methyl benzylamine. This suggests that zinc could be an important risk factor with great therapeutic value in ESCC.
Based on the candidate genes, their interactions and the toxicogenomics network we present here, gives the opportunity to formulate several important questions towards the investigation of ESCC. Such as, the mode of action of risk factors and drug resistance [71, 72]. For instance, toxicogenomic inferences of differential genes have shown Tobacco smoke pollution and cisplatin share a larger number (
Among the candidate genes, the survival analysis showed STC2 (Stanniocalcin-2) gene has a risk effect on survival of ESCC patients. This gene is shown to play a critical role in calcium-phosphate homeostasis and is seen overexpressed in a broad spectrum of tumor cells [74, 75] including esophagus. Under stress conditions like ER stress, hypoxia and nutrient deprivation it gets stimulated and promotes cell proliferation, migration and immune response. Further, known to promote the development of acquired resistance to chemo- and radio-therapies [76]. Hence, STC2 is a potential cross-cancer biomarker and a therapeutic target.
There are some potential limitations to consider in this study. Due to our use of a meta-analysis approach, we were only able to examine the common genes (5080) found in the datasets for the differential expression (DE) analysis. Unfortunately, a significant number of genes had to be excluded from the analysis due to variations in gene coverage among different technology platforms. Consequently, we were unable to identify potential post-transcriptional modifications or translational end products for many genes using ESCC ATLAS.
Conclusion
In this contribution, using the mRNA expression profiles of publicly available 10 datasets (together comprising 283 tumor and 290 normal quality samples), we highlight molecular connections to microbiota of esophagus, environmental chemical exposures, and (deficiency of) trace element zinc in conjunction with core genes led adjoining gene perturbation in ESCC etiology, and gene interaction with therapeutic agents. Our attempt of functional enrichment of the candidate genes deciphered crucial ‘bittersweet’ biological pathways which can be exploited for the ESCC treatment.
Availability of data and materials
The data generated in this study are available within the article and its supplementary data files.
Funding
This research project was funded by the Deanship of Scientific Research, Princess Nourah bint Abdulrahman University, through the Program of Research Project Funding After Publication, grant No (43- PRFA-P-8)
Authors’ contributions
P.H, A.A, and A.B conceptualized the study. V.P.G, P.S.G, S.R, and P.H performed data analysis, V.P.G, J.P and P.H did interpretation the analysis results. V.P.G, and P.H wrote the manuscript. J.P, R.N.S, S.M, D.M, A.T, L.B.V, A.K.B and M.W.C were involved in revision for important intellectual content of the manuscript. A.A, A.B and PH supervised the research work. All authors have reviewed and approved the final manuscript.
Supplementary data
The supplementary files are available to download from
sj-docx-1-cbm-10.3233_CBM-230145.docx - Supplemental material
Supplemental material, sj-docx-1-cbm-10.3233_CBM-230145.docx
sj-xlsx-1-cbm-10.3233_CBM-230145.xlsx - Supplemental material
Supplemental material, sj-xlsx-1-cbm-10.3233_CBM-230145.xlsx
sj-xlsx-2-cbm-10.3233_CBM-230145.xlsx - Supplemental material
Supplemental material, sj-xlsx-2-cbm-10.3233_CBM-230145.xlsx
Footnotes
Conflict of interest
Authors do not have any competing interests.