
Abstract
BACKGROUND:
Lymphoid-specific helicase (HELLS), a SNF2-like chromatin-remodeling enzyme, plays a key role in tumor progression via its DNA methylation function. However, the effects of HELLS on immune infiltration and prognosis in liver hepatocellular carcinoma (LIHC) remain uncertain.
METHODS:
The Tumor Immune Estimation Resource (TIMER) database was employed to explore the pan-cancer mRNA expression of HELLS and its correlation with immunity. GEPIA2 was used to verify the correlation between HELLS expression and survival. The role of HELLS in cancer was explored via gene set enrichment analysis (Gene Ontology and Kyoto Encyclopedia of Genes and Genomes) and the construction of gene-gene and protein-protein interaction networks (PPI). Additionally, correlations between DNA methylation, HELLS expression, and immune-related genes were explored in LIHC. HELLS expression in LIHC clinical samples was determined using qRT-PCR and western blotting. The effects of downregulated HELLS expression in hepatocellular carcinoma cells was explored via transfection experiments in vitro.
RESULTS:
High HELLS mRNA expression was identified in several cancers and was significantly associated with poorer prognosis in LIHC. Furthermore, HELLS expression was positively correlated with tumor-infiltrating lymphocytes and immune checkpoint genes in LIHC. Bioinformatics analysis suggested that DNA methylation of HELLS may be associated with the immune response. Results from the TCGA-LIHC dataset, clinical samples, and functional analysis indicated that HELLS contributed to tumor progression in LIHC.
CONCLUSION:
The study findings demonstrate that HELLS is an important factor in promoting LIHC malignancy and might serve as a potential biomarker for LIHC.
Introduction
Cancer incidence and mortality are booming worldwide [1]. The incidence of gastrointestinal and hepatic cancers continues to increase rapidly, which has also contributed to the continuing increase in cancer-related deaths [2]. Liver hepatocellular carcinoma (LIHC), the most common subtype of hepatic cancer, ranked third in tumor-related mortality worldwide. Although some patients with early LIHC can be cured surgically, approximately 70% of patients relapse after five years [3]. Meanwhile, increasing evidence indicates that immunotherapy is a potential prospect for LIHC treatment [4].
Lymphoid-specific helicase (HELLS, also known as LSH) is a 97 kDa transmembrane protein found in many cell types and organs. HELLS is important for normal embryonic development because it interacts with DNA methyltransferases [5]. Strikingly, accumulating evidence has identified HELLS as an oncogenic molecule that promotes tumorigenicity. In addition, scattered evidence has also suggested that HELLS is needed for malignant progression and stem cell maintenance [6, 7]. More significantly, the prognosis of patients is not optimistic for those with upregulated HELLS expression. Nevertheless, the underlying mechanism of HELLS in tumor progression remains unclear.
DNA methylation, chromatin structure regulation, and histone post-translational modifications, which are epigenetic mechanisms, are critical for interactions between tumor and immune cells [8]. Immune-related mechanisms are key points in hepatic cancers, while at the same time, immunotherapeutic strategies are regarded as a promising direction for the treatment of hepatic cancers [9]. Cancer immunotherapy has attracted significant attention, and treating LIHC with immunotherapy is a tantalizing prospect. DNA methylation and gene expression have potential complementarity in the prediction of immune responses [10]. Therefore, the potential correlation between immunity and DNA methylation in LIHC caught our attention.
In this study, we assessed the expression signature and survival value of HELLS gene in pan-cancer by Tumor Immunity Estimation Resource (TIMER) and TCGA database. we evaluated the potential correlation between HELLS and tumor-infiltrating lymphocytes in LIHC. In addition, the correlation of DNA methylation and HELLS in LIHC were also evaluated by bioinformatics. Finally, we verified the HELLS expression in LIHC by clinical LIHC samples. According to these results, it is suggested that HELLS could make a difference in prognosis of LIHC by affecting tumor immunity, and on this basis, HELLS could be a new promising biomarker for LIHC immunotherapy.
In the current study, we hypothesized that HELLS may be an important biomarker in LIHC and contribute to tumor immunity. Thus, the aims of the study were to: 1) assess the pan-cancer expression signature and survival value of HELLS; 2) evaluate the potential correlation between HELLS expression and tumor-infiltrating lymphocytes in LIHC; 3) explore the correlation between DNA methylation and HELLS expression in LIHC; 4) verify HELLS expression in clinical LIHC samples. The study findings may provide a promising new promising biomarker for LIHC immunotherapy.
Methods
TIMER database analysis
TIMER (
GEPIA2 analysis
Survival analysis was performed using Gene Expression Profiling Interactive Analysis (GEPIA2) (
LinkedOmics database analysis
Gene expression profiles were investigated using the LinkedOmics platform (
Gene interaction analysis
The GeneMania database (
DNA methylation-related database analysis
The Molecular Signatures Database (MSigDB) (
The clinical characteristics of patients in the TCGA dataset
The clinical characteristics of patients in the TCGA dataset
HELLS expression and clinical data were obtained from the TCGA-LIHC dataset, including 374 tumor samples and 50 normal tissue samples. Cancerous subtype in the TCGA-LIHC dataset was defined using non-negative matrix factorization (NMF) with the “cancertypes” packages in R software (version 3.63). Table 1 lists the relevant information of clinical samples obtained from the TCGA-LIHC dataset.
Sample collection
To further verify HELLS expression in LIHC, samples of tumors and matched normal tissues were obtained from 8 patients with primary hepatocellular carcinoma (HCC). The samples were collected at the First Affiliated Hospital of Kunming Medical University. Signed informed consent was provided by patients or their family members. This study was approved by the Ethics Committee of the First Affiliated Hospital of Kunming Medical University.
qRT-PCR
qRT-PCR was performed as previously described[21]. gDNA eraser and cDNA was synthesized using reverse transcription with the PrimeScript RT Reagent Kit (TaKaRa, Kusatsu, Japan). PCR was performed using the following specific primers: HELLS-Forward 5
Western blotting
Western blotting was performed as previously described [22]. HELLS rabbit polyclonal antibody (11955-1-AP) and GAPDH (10494-1-AP) were purchased from Proteintech (Wuhan, China). The membranes were detected by using AI600 (GE Healthcare Life Sciences, America) and densitometric analyses by ImageJ software.
Cell culture and transfection
The LIHC line SKHep-1, Huh7, SNU-387 were obtained from Pricella (Wuhan, China). Dulbecco’s Modified Eagle’s medium (Gibco, USA) containing 10% fetal bovine serum and 1% penicillin streptomycin (Gibco) was used to culture the cells. The cells were incubated at 37
Short hairpin RNA (shRNA) was constructed to regulate HELLS expression. The shRNAs targeting HELLS were designed by Shanghai Genechem (Shanghai, China) as follows: shRNA-1:5
Cell migration
For the cell migration studies, SKHep-1 cells (5
Wound healing assay
SKHep-1 cells were cultured to 90% confluence in 6-well plates. A sterile 200
CCK-8 assay
SKHep-1 cells (5
EDU assay
SKHep-1 cells were cultured in 24-well plates and cell proliferation was measured using the Click-iT
Statistical analysis
GraphPad Prism (Ver. 8.0.1) was performed to statistical analysis and presented as mean
Results
Pan-cancer HELLS expression and prognosis analysis
To investigate differential HELLS expression in multiple types of cancer, HELLS mRNA levels were analysed in tumor and adjacent normal tissues of different cancer types in the TIMER database (Fig. 1A). The results indicated that HELLS mRNA expression differed significantly between tumor and adjacent normal tissues in 19 cancer types. Additionally, HELLS mRNA expression was inconsistently higher or lower in diverse cancer types. For example, HELLS mRNA levels were significantly higher in bladder urothelial carcinoma (BLAC), breast invasive carcinoma (BRCA), cholangial carcinoma (CHOL), colon adenocarcinoma (COAD), esophageal carcinoma (ESCA), head and neck squamous cell carcinoma (HNSC), kidney chromophobe (KICH), LIHC, lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), prostate adenocarcinoma (PRAD), rectum adenocarcinoma (READ), stomach adenocarcinoma (STAD), thyroid carcinoma (THCA), and uterine corpus endometrial carcinoma (UCEC). In contrast, HELLS mRNA levels were significantly lower in kidney renal clear cell carcinoma (KIRC) and skin cutaneous melanoma (SKCM).
Next, GEPIA2 was employed to obtain an in-depth understanding of the correlation between HELLS expression and prognosis in cancers which all Fig. 1A types. (Fig. 1B and C) Higher HELLS expression was correlated with worse overall survival (OS) (
HELLS mRNA expression and prognostic significance in multiple types of cancers. A. HELLS mRNA levels in different tumor types from the TIMER database. 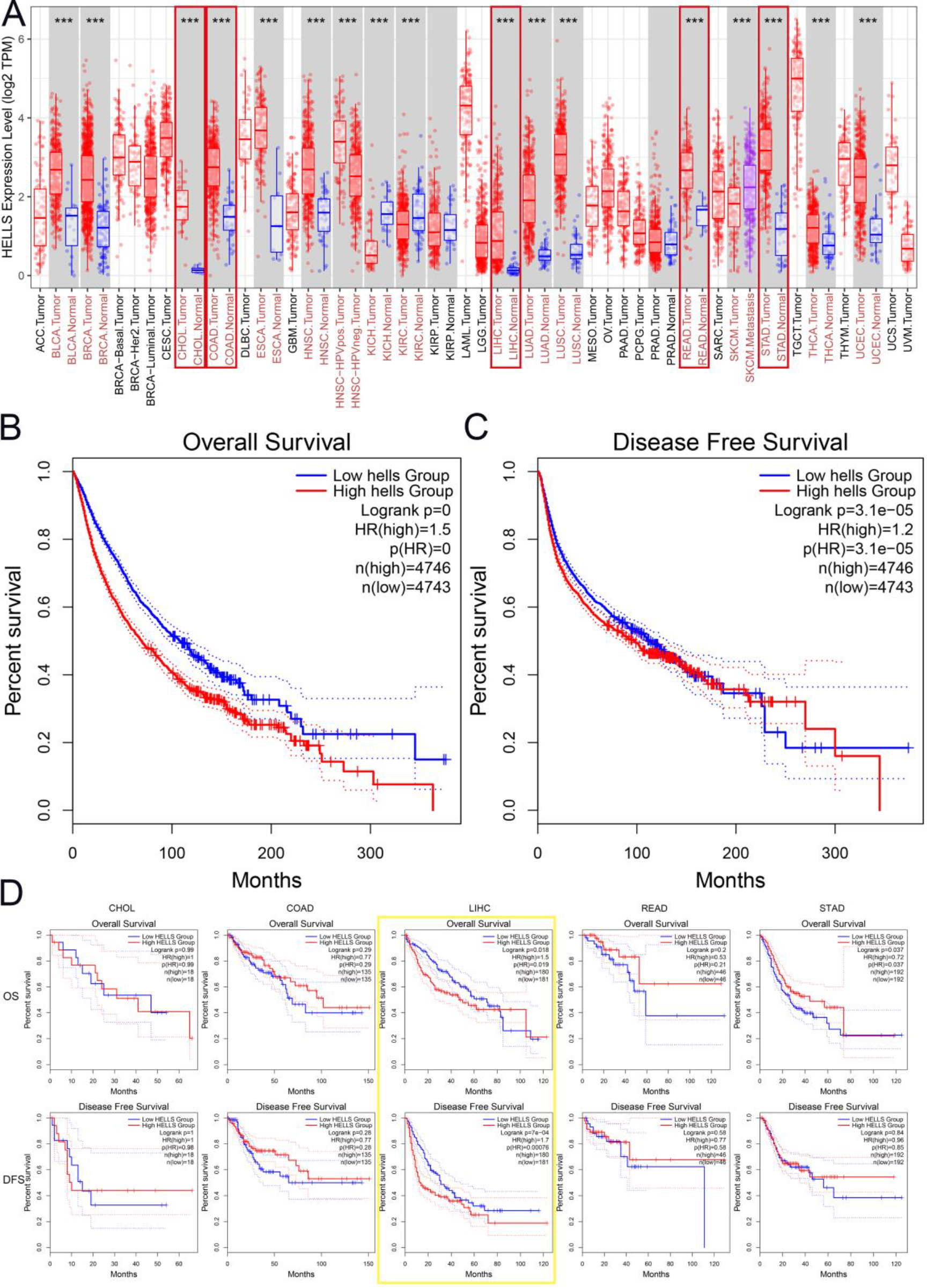
Notably, HELLS expression was significantly higher in gastrointestinal and hepatic cancers in the TIMER database. Thus, RNA sequencing data from the TCGA dataset were analyzed to determine the prognostic potential of HELLS in gastrointestinal and hepatic cancers via GEPIA2. Surprisingly, higher HELLS expression was correlated with significantly poorer OS (HR
Tumor-infiltrating lymphocytes are known to be closely related to tumor grade, TNM stage, and prognosis, especially in gastrointestinal and hepatic cancers [23, 24]. Thus, the correlation between HELLS expression and immune cell infiltration in gastrointestinal and hepatic cancers was investigated in the TIMER database (Fig. 2A). The results showed that high HELLS expression was highly positively correlated with immune cell infiltration in gastrointestinal and hepatic cancers. HELLS expression was positively correlated with levels of infiltrating CD8
Next, correlations between HELLS expression and ICP genes among gastrointestinal and hepatic cancers were explored (Supplementary Fig. 1). Among 72 ICP genes, strong correlations with HELLS expression were identified in LIHC. HELLS expression was positively correlated with 59 of 72 ICP genes (82%) in the non-purity group (Supplemental Table 1). Moreover, in LIHC, 66 of 72 ICP genes (92%) were positively correlated with HELLS expression in the purity group. However, HELLS expression was weakly correlated with ICP genes in COAD, CHOL, READ, and STAD.
Subsequently, correlations between HELLS expression and PD-L1, CTLA4, and PD-1 were explored in gastrointestinal and hepatic cancers using ENCORI (Fig. 2B). HELLS expression was positively correlated with PD-L1 (
Correlations between HELLS expression, tumor-infiltrating lymphocytes, and ICP genes in gastrointestinal and hepatic cancers by TIMER. A. Correlations between HELLS expression and different tumor-infiltrating lymphocytes. B. Correlations between HELLS expression and ICP genes.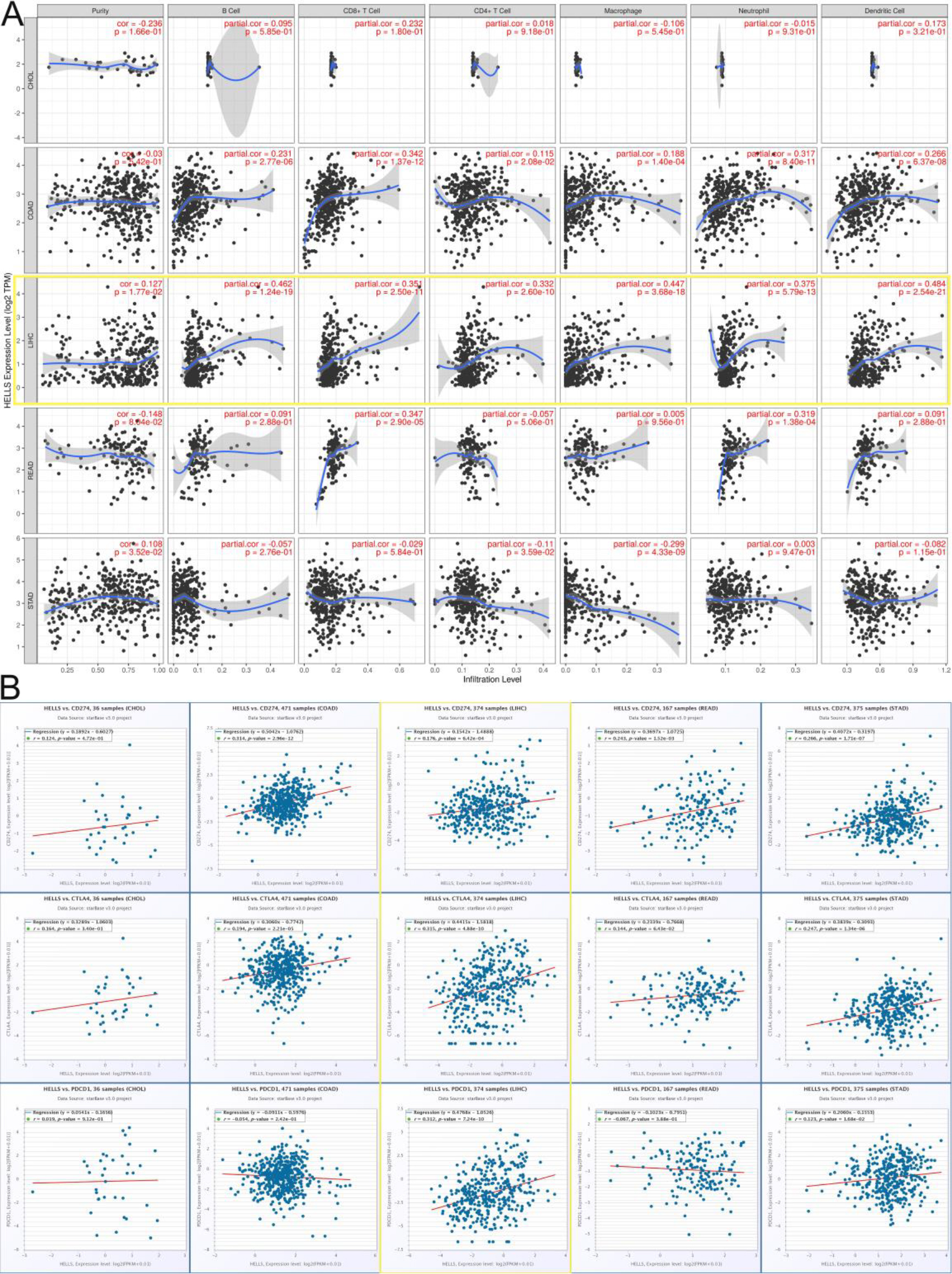
Finally, correlations between HELLS expression, tumor-infiltrating lymphocytes, and ICP genes in gastrointestinal and hepatic cancers were analysed. HELLS upregulation was correlated with increased immune-cell infiltration, suggesting that HELLS expression could have a relationship with the tumor immune microenvironment (TME), especially in LIHC. Thus, these results further confirmed that HELLS expression was correlated with infiltrating immune cells in LIHC.
Correlations between HELLS expression and genes in LIHC analysed using the LinkedOmics database. A. Top 50 genes positively correlated genes with HELLS in LIHC. B. Top 50 genes negatively correlated with HELLS in LIHC. C. Top 20 GO molecular function terms associated with HELLS-related genes in LIHC. D. KEGG pathway enrichment analysis of HELLS-related genes in LIHC. E. Gene-gene interaction network for HELLS in LIHC. F. Protein-protein interaction network for HELLS in LIHC. G. GO enrichment analysis of HELLS-interacting genes in LIHC.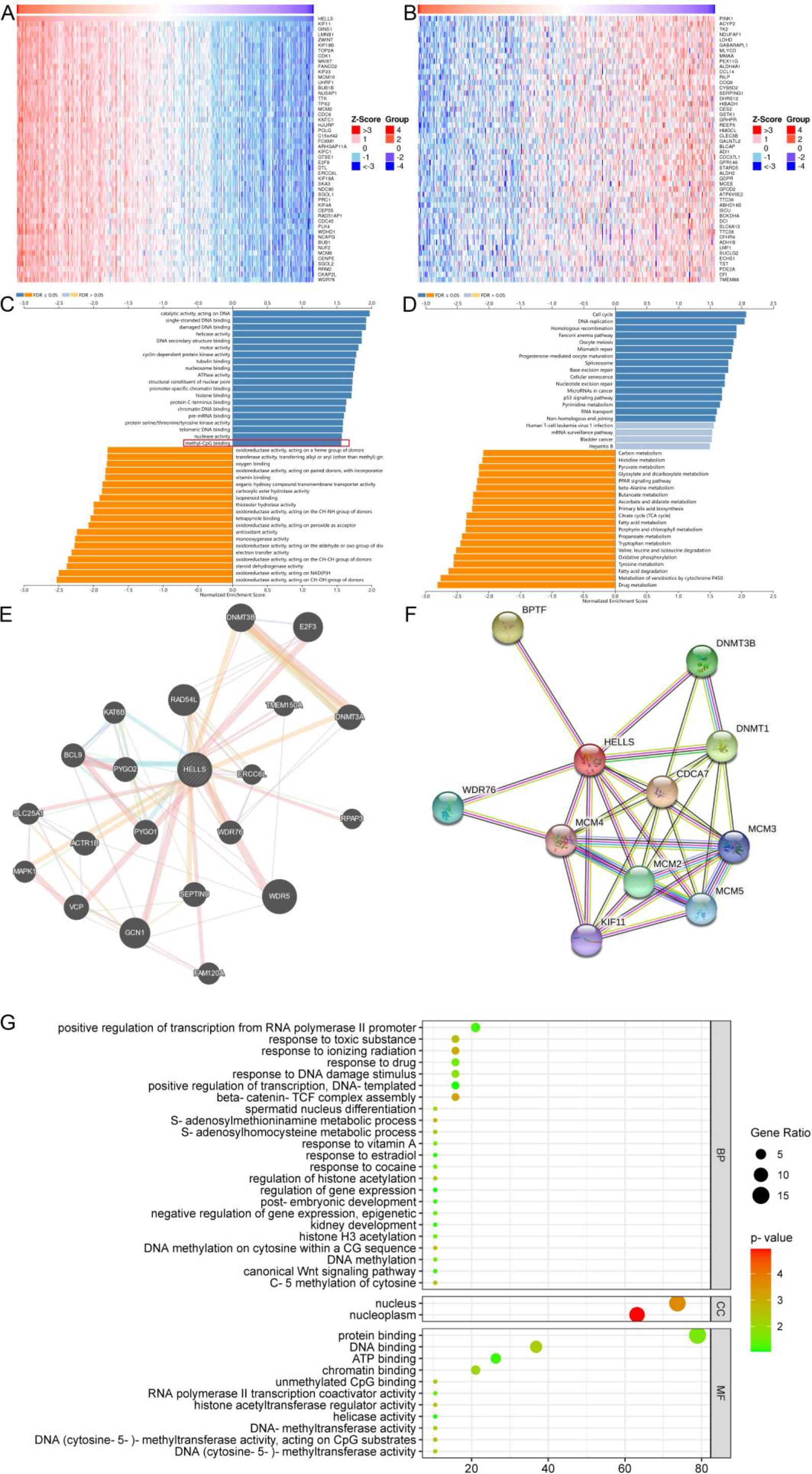
GSEA was performed to help elucidate the molecular mechanisms affected by HELLS in LIHC using data from the TCGA. Heat maps were used to illustrate the top 50 co-expressed genes that were positively and negatively correlated with HELLS in LIHC (Fig. 3A, B). Genes highly correlated with HELLS were determined by Pearson’s correlation analysis in the LIHC cohort (Supplementary Fig. 2A). Next, GO and KEGG enrichment analyses were used to explore the functions and signaling pathways related to HELLS in LIHC.
Hepcidin-related pathways and biological functions were analysed for 300 genes that were positively correlated with HELLS via KEGG and GO enrichment analyses. The top 20 significant GO biological process (BP) terms (Supplementary Fig. 2B), cell component (CC) terms (Supplementary Fig. 2C), and molecular function (MF) terms (Fig. 3C), as well as KEGG enriched pathways (Fig. 3D), were presented. Notably, in terms of GO MFs, co-expressed genes were enriched in DNA-methylation related functions such as histone binding, methyl-CpG binding, etc. In terms of KEGG pathways, co-expressed genes were enriched in pathways associated with the cell cycle, DNA replication, and cancer-related pathways. These results indicated that HELLS expression might be involved in regulating the immune response in LIHC via DNA-related functions.
The GeneMania database was searched to construct the gene-gene interaction network for HELLS. The network indicated that the 20 most frequently alteredgenes, including DNA methyltransferase 3A (DNMT3A) and DNA methyltransferase 3B (DNMT3B), were closely correlated with HELLS expression (Fig. 3E). These genes were chosen for GO enrichment analysis (Fig. 3G), which identified many DNA methylation-related GO terms, such as DNA methylation and DNA methylation activity. Subsequently, a PPI network was constructed for HELLS using the STRING database (Fig. 3F). The PPI network included 11 nodes and 33 edges, which contained DNA-methyltransferase 1 (DNMT1) and DNMT3B. The results suggested that DNA methylation may play a key role of HELLS in LIHC.
Correlation between HELLS expression and DNA methylation
Tumor-infiltrating lymphocytes and DNA methylation are regarded as key influencing factors of tumor progression [25]. MSigDB was used to investigate the correlation between HELLS expression and DNA methylation. GO BP terms, including maintenance of DNA methylation (GO-0010216) and DNA methylation-dependent heterochromatin assembly (GO-0006346) were identified, and gene correlation analysis was performed via GEPIA2 (Fig. 4A, B). CTCF, DNMT1, UHRF1, UHRF2, USP7, and ZNF445 were positively correlated with HELLS expression in GO-0010216. Similarly, AICDA, APOBEC1, ATF7IP, DNMT3A, HDAC1, MBD2, MBD3, MORC2, MPHOSPH8, PPHLN1, SETDB1, SETDB2, SIRT1, TET1, TRIM28, and ZNF304 were positively associated with HELLS expression in GO-0006346. HELLS might be closely related to DNA methylation in HCC and might affect the occurrence and development of HCC through DNA methylation. At present, DNA methylation markers are widely associated with differential cancer survival. Thus, promoter methylation expression was analysed using UALCAN (
The information of clinical samples (
8)
The information of clinical samples (
TNM is the most widely used tumor clinical staging system, with T representing the size of the tumor. Tumor grade assesses the pathologic degree of the tumor tissue, including the number of nuclear divisions, arrangement, degree of differentiation, and local infiltration of cancer cells. Tumor status is used to describe whether the tumor has spread. The expression of HELLS in LIHC at different TNM stages, pathologic stages, tumor status, alpha-fetoprotein (AFP) levels, and age was analysed in the TCGA dataset using R software (Table 1). Compared with normal tissues, HELLS expression was significantly higher in LIHC tissues (Fig. 4E). HELLS expression was significantly higher in stages T1
Analysis of HELLS DNA methylation and expression in LIHC. A. Co-expression analysis of the GO term maintenance of DNA methylation (GO-0010216) in LIHC by MSigDB. B. Co-expression analysis of the GO term DNA methylation-dependent heterochromatin assembly (GO-0006346) in LIHC by MSigDB. C. Promoter methylation levels of HELLS in LIHC by UALCAN. D. ROC analysis of HELLS expression in LIHC based on the TCGA dataset. E. Correlation between HELLS expression in normal and LIHC samples based on the TCGA dataset. F. Correlation between HELLS expression and tumor stage (T) in LIHC based on the TCGA dataset. G. Correlation between HELLS expression and pathologic stage of patients with LIHC based on the TCGA dataset. H. Correlation between HELLS expression and tumor status in LIHC based on the TCGA dataset. I. Correlation between HELLS expression and alpha-fetoprotein (AFP) level in LIHC based on the TCGA dataset. J. Correlation between HELLS expression and age in LIHC based on the TCGA dataset. K. Correlation between HELLS expression and number of affected lymph nodes (N) in LIHC based on the TCGA dataset.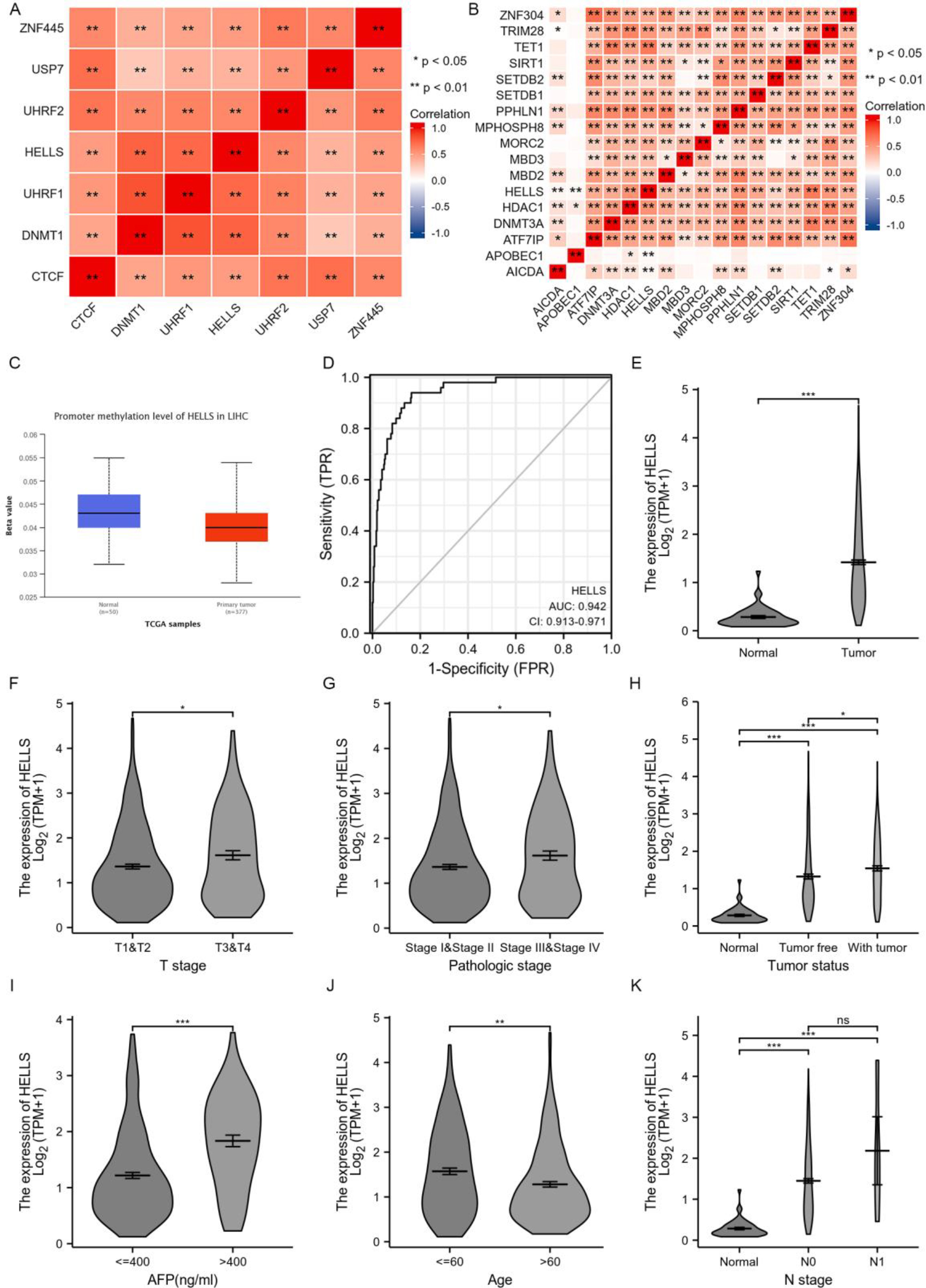
Downregulated of HELLS expression suppressed LIHC growth and migration (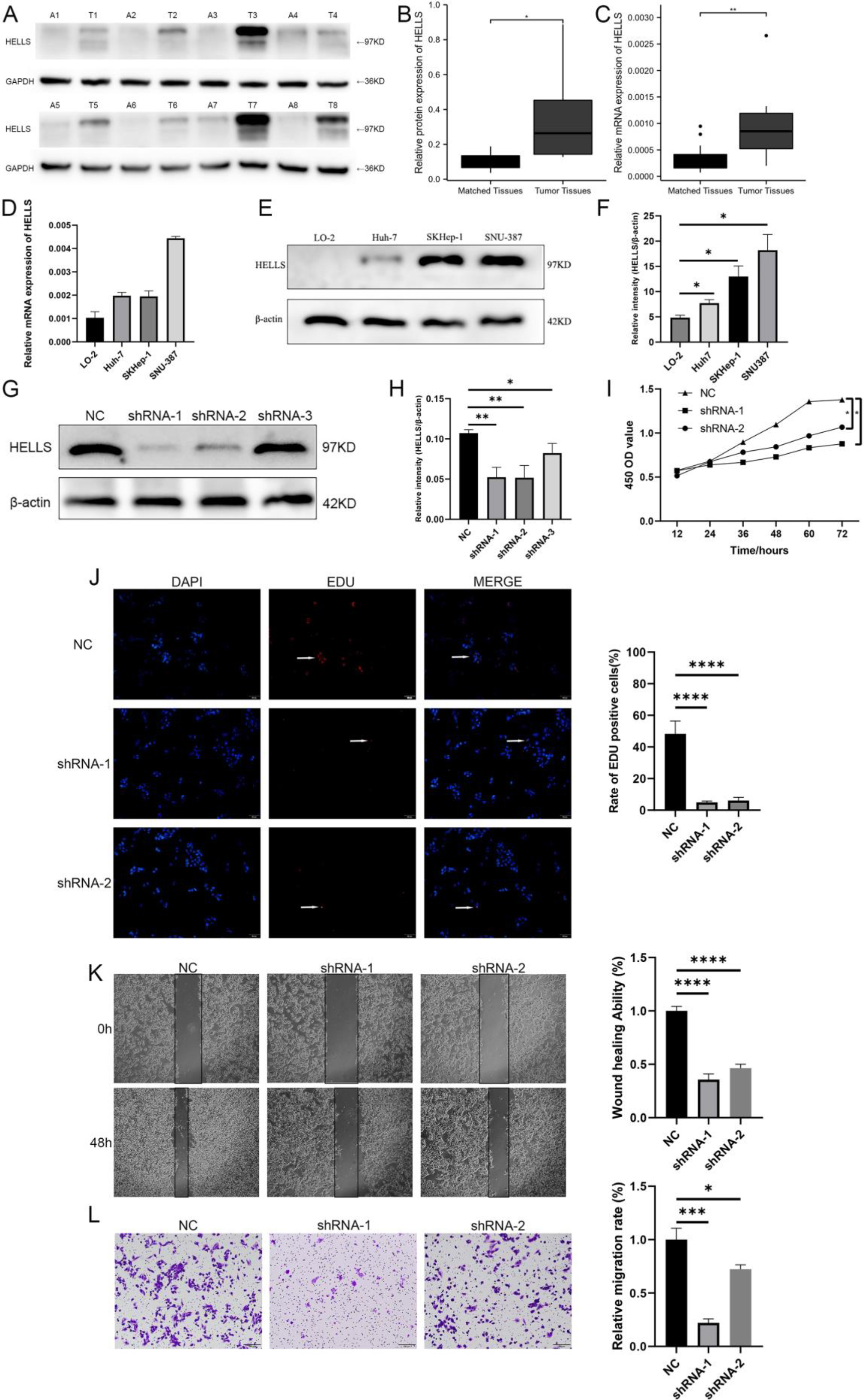
To validate HELLS expression in clinical samples, HELLS protein and mRNA levels were assessed in matched tumor and normal tissues of an HCC cohort (
Effect of HELLS knockdown on LIHC malignant behaviors
According to the results, HELLS expression was higher in tumor tissues than in matched normal tissues. Additionally, HELLS mRNA and protein levels were increased in LO2, Huh7, SKHep-1, and SNU-387 tumor cells. Compared with LO-2, the level of mRNA and protein differences of Huh7, SKHep-1, and SNU-387 were statistically significant (
Discussion
HELLS, a DNA helicase related to the SWI/SNF2 family, affects DNA methylation and chromatin packaging. HELLS has been shown to promote tumor development and progression in various cancers, as well as the proliferation and invasion of cancer cells [26]. In spite of the key roles played by HELLS in cancer, HELLS has not been sufficiently studied in the context of immunity and oncology. Therefore, this study explored the pan-cancer expression of HELLS and revealed its potential immunological role in gastrointestinal and hepatic cancers via bioinformatics analysis.
The initial investigation revealed that HELLS expression was higher in different malignancies than in normal tissues and was positively correlated with lower survival rates in patients. The high HELLS expression found in various types of tumors was consistent with the results reported in other recent studies [27, 28]. Notably, HELLS expression was correlated with the poorest prognosis in LIHC, which was comparable with gastrointestinal tumors. These results indicated that HELLS promotes tumor progression and development in malignancies, and may provide a potential prognostic biomarker in LIHC.
The immune system has great significance in the monitoring and clearing of tumor cells [29]. Therefore, pan-cancer HELLS expression was investigated in various immune-related molecular subtypes to determine the potential underlying mechanism. HELLS expression was correlated with a variety of immune cells in gastrointestinal and hepatic cancers, especially in LIHC. HELLS expression was positively correlated with levels of CD8
Further exploring the functions and signaling pathways affected by HELLS in LIHC through GSEA revealed that HELLS was mainly associated with DNA-related functions such as DNA methylation. In the constructed gene-gene and protein-protein interaction networks, some notable genes and proteins interacted with HELLS, including DNMT1, DNMT3A, and DNMT3B. These results suggest that HELLS and its interacting genes and proteins may take part in regulating the immune response via DNA methylation in LIHC. Previous studies have reported that HELLS is a key epigenetic driver of cancers[26, 37]. Therefore, the correlation between HELLS expression and DNA methylation was explored in the current study. Unsurprisingly, the MSigDB data mining results revealed that HELLS was associated with GO terms such as maintenance of DNA methylation and DNA methylation-dependent heterochromatin assembly. Meanwhile, Han et al. provided evidence verifying that HELLS drives DNA methylation via DNMT1, and downregulation of HELLS contributed more to DNA methylation than downregulation of DNMT3A and DNMT3B [38]. Moreover, Termanis et al. reported that the ATP-binding function of HELLS was important for cell-autonomous de novo DNA methylation [39]. HELLS expression was negatively correlated with DNA methylation in the current study. Thus, we hypothesize that HELLS, as a methylation gene, may regulate the methylation of other DNA methylation-related genes in LIHC.
Recent seminal studies have discovered that HELLS expression is upregulated in several types of tumors, such as prostate cancer, LUAD, and colorectal cancer [7, 40, 41, 42]. and HELLS is probably involved in tumor progression. In the current study, drastic upregulation of HELLS expression was verified in LIHC via bioinformatics analysis and in clinical samples, and HELLS expression was remarkably correlated with TNM stage, pathologic stage, tumor status, AFP, and age in patients with LIHC. Additionally, high HELLS expression was significantly correlated with worse OS and DFS in LIHC. These findings highlight the potential significance of HELLS upregulation in LIHC progression and prognosis. Further, the transfection experiments confirmed that downregulation of HELLS could inhibit the proliferation and migration of HCC cells.
In summary, HELLS was upregulated in gastrointestinal tumors and LIHC. HELLS expression was closely correlated with poor prognosis and levels of infiltrating CD8
Conclusion
This study found abnormal HELLS expression in a variety of tumors, including LIHC, which was significantly associated with poor prognosis in LIHC. The correlation between HELLS expression and immune cell infiltration was demonstrated via bioinformatics analysis and further validated in clinical samples using western blotting and qRT-PCR. Targeting HELLS may provide a new direction for the clinical diagnosis and treatment of LIHC. Nevertheless, given the unclear mechanism underlying the role of HELLS in the occurrence and development of LIHC, further studies are warranted to elucidate the impact of HELLS and its epigenetics on the TME of LIHC.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the First Affiliated Hospital of Kunming Medical University.
Consent for publication
All authors have read and agreed to the published version of the manuscript.
Availability of data and material
The data used to support the findings of this study are available from the corresponding author upon request.
Funding
National Natural Science Foundation of China (No. 81960124). Funded by the Scientific Research Fund of the Education Department of Yunnan Province (2022Y199).
Author contributions
Conceptualization and financial support: HanFei Huang, YingLei Miao and Zhong Zeng.
Analyzed the data, carried out the main experiments, and drafted this manuscript: Yuan Fang, WeiQiang Tang and Dan Zhao. These authors contributed equally to this work.
Technical support and manuscript revision: XiaoLi Zhang, Na Li, Yang Yang, Li Jin, ZhiTao Li and BenKai Wei.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-230073.
sj-docx-1-cbm-10.3233_CBM-230073.docx - Supplemental material
Supplemental material, sj-docx-1-cbm-10.3233_CBM-230073.docx
Footnotes
Acknowledgments
Not Applicable.
Conflict of interest
The authors declare no conflict of interest.