
Abstract
BACKGROUND:
Despite its importance in the clinical management of ovarian cancer, the CA125 biomarker – located on the mucin protein MUC16 – is still not completely understood. Questions remain about MUC16’s function and structure, specifically the identity and location of the CA125 epitopes.
OBJECTIVE:
The goal of this study was to characterize the interaction of individual recombinant repeats from the tandem repeat domain of MUC16 with antibodies used in the clinical CA125 II test.
METHODS:
Using E. coli expression, we isolated nine repeats from the putative antigenic domain of CA125. Amino acid composition of recombinant repeats was confirmed by high-resolution mass spectrometry. We characterized the binding of four antibodies – OC125, M11, “OC125-like,” and “M11-like” – to nine recombinant repeats using Western blotting, indirect enzyme-linked immunosorbent assay (ELISA), and localized surface plasmon resonance (SPR) spectroscopy.
RESULTS:
Each recombinant repeat was recognized by a different combination of CA125 antibodies. OC125 and “OC125-like” antibodies did not bind the same set of recombinant repeats, nor did M11 and “M11-like” antibodies.
CONCLUSIONS:
Characterization of the interactions between MUC16 recombinant repeats and CA125 antibodies will contribute to ongoing efforts to identify the CA125 epitopes and improve our understanding of this important biomarker.
Introduction
Considerable effort has been directed toward expanding the suite of biomarkers available for diagnosing and monitoring high-grade serous ovarian cancer (HGSOC) [1, 2, 3, 4, 5, 6, 7, 8]. Although new biomarkers – most significantly human epididymis protein 4 (HE4) [9, 10] – have been identified and are proving to be transformative, enabling new assays and algorithms [11, 12, 13, 14, 15, 16], no biomarker has supplanted cancer antigen 125 (CA125), which remains the clinical gold standard for monitoring response to treatment and detecting cancer recurrence [17, 18, 19, 20]. The FDA-approved assay for CA125 is widely used, despite the fact that the CA125 epitopes have not been defined and controversy persists regarding the minimal functional unit necessary for antibody detection [21, 22, 23]. In other words, the test that underlies vital decisions in ovarian cancer care employs a mechanism that is not understood.
The CA125 epitopes are carried on MUC16, a large mucin [24, 25]. Some structural features of MUC16 have been determined, including a highly glycosylated N-terminal domain, an immunologically active tandem repeat region containing many similar but non-identical subdomains, and a C-terminal domain including a transmembrane region and cytoplasmic tail [26, 27]. The circulating form of MUC16 detected with the CA125 II ELISA does not contain the transmembrane or cytoplasmic regions. Based on competition studies, CA125 antibodies have been sorted into three groups: OC125-like (group A), M11-like (group B), and OV197-like (group C) [28, 29]. The CA125 II ELISA test is a double determinant immunoassay using two different antibodies (M11 and OC125) as capture and tracer [30]. Earlier iterations of the assay used OC125 as both capture and tracer [31]. The location and identity of the CA125 epitopes remain undefined, and diverse experimental approaches to determine the epitopes have been reported [32, 33, 34, 35, 36]. Experiments by Bressan and co-workers using Western blot analysis of six recombinantly expressed repeat domains (R2, R7, R9, R11, R25, and R51) revealed that the antibodies used in the CA125 II test (OC125 [31] and M11 [37]) do not recognize all repeat domains uniformly [35]. We hypothesized that variation in antibody recognition may be observed in other recombinant repeats and that the nature of the molecular recognition assay may influence whether binding is observed, particularly if the CA125 epitope is conformational [38].
Here we report the expression of nine recombinant repeats from the putative antigenic domain of CA125 (R2, R5, R6, R7, R9, R11, R25, R34, and R58, in the O’Brien numbering system, sequence alignment shown in SI Fig. 1) [26]. Using Western blotting, indirect ELISA, and localized surface plasmon resonance (SPR) spectroscopy, we characterized the interactions of expressed and purified recombinant repeats with four CA125-binding monoclonal antibodies (OC125, M11, “OC125-like,” and “M11-like”). Consistent with our hypothesis and previous reports, the epitopes were found to be distributed nonuniformly over the recombinant repeats, and variation across assay method was observed. Without knowledge of the CA125 epitopes, it is impossible to characterize individual variation in MUC16 proteoforms or to determine whether MUC16 expression changes during cancer development, in response to treatment, or during recurrence. To improve the long-term survival of ovarian cancer patients, there is a vital need to improve the diagnostic value of CA125. This study represents the largest set of MUC16 recombinant repeats, the largest number of antibodies, and the most molecular interaction assay methods reported to date and contributes to ongoing efforts to understand the molecular nature and immunological activity of CA125. The rationale of this study is that its results will ultimately enable us to reinvent the CA125 test by developing new affinity reagents to supplement or replace the antibodies in current use, since a biomarker is only as good as the tools available to detect it.
Materials and methods
Recombinant repeat expression and purification
The MUC16 coding sequence as described by O’Brien and co-workers [26] was obtained from NCBI (GenBank: AF414442.2). The nucleotide sequences of nine tandem repeats were synthesized and cloned into pET14b vector (GenScript, Piscataway, NJ), which was used to express protein with N’ 6xHistidine-tagging (6xHis) by XhoI and BamHI sites. Each plasmid was transformed into the SHuffle T7 Express E. coli strain (New England Biolabs, Beverly, MA). Bacteria clones were grown in Luria-Bertani (LB) broth (Thermo, Waltham, MA) containing 100
Mass spectrometry
The bottom-up proteomics workflow, including sample preparation and liquid chromatography-tandem mass spectrometry (LC-MS/MS), closely followed our previously published reports [39, 40]. Briefly, 10
Western blot
Recombinant repeats were resolved by 16% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 5% non-fat milk in TBS-T buffer (Tris buffered saline with 0.05% Tween 20) at RT for 1 hour. The membrane was hybridized at 4
ELISA
Pierce Nickel-Coated Plates (Thermo) were used to immobilize recombinant repeats through their 6xHis tag for indirect ELISA. Five hundred ng of recombinant repeat in 100
Surface plasmon resonance
All SPR experiments were performed on a Nicoya Lifesciences OpenSPR-XT 2-channel instrument (Kitchener, ON) using Nicoya NTA-functionalized standard sensor chips. PBS with 0.05% Tween 20 (PBS-T; PBS from GrowCells, Irvine, CA; Tween 20 from Fisher Scientific, Waltham, MA) was used as both the immobilization and running buffer. Autosampler tray and sensor temperatures were set to 20
After surface regeneration with EDTA, the surface was activated with 40 mM NiCl
Recombinant repeats expressed and characterized in this study. Repeat numbering, amino acid sequences and corresponding amino acid number in reference sequence AF414442.2, as reported by O’Brien and co-workers [26], are shown in left three columns. The SEA domain extends from repeat 1 to 126 and is followed by a region rich in proline, serine, and threonine. Repeats with underlined numbers were previously examined by Bressan et al. [35]. Tandem repeat numbering increases from N- to C-terminus. Peptide coverage determined from bottom-up proteomics experiments to confirm expression is shown in the right column
Recombinant repeats expressed and characterized in this study. Repeat numbering, amino acid sequences and corresponding amino acid number in reference sequence AF414442.2, as reported by O’Brien and co-workers [26], are shown in left three columns. The SEA domain extends from repeat 1 to 126 and is followed by a region rich in proline, serine, and threonine. Repeats with underlined numbers were previously examined by Bressan et al. [35]. Tandem repeat numbering increases from N- to C-terminus. Peptide coverage determined from bottom-up proteomics experiments to confirm expression is shown in the right column

Western blot images of recombinant repeats from MUC16 probed with antibody clones OC125 (1:200), M61704 (OC125-like, 1:2000), M11 (1:100), and M61703 (M11-like, 1:2000), and an anti-His (His.H8, 1:2000) loading control.
Recombinant tandem repeat protein expression and confirmation
Recombinant repeats were expressed in E. coli and purified on Ni-NTA beads. Yield and purity of material covered from Ni-NTA beads were confirmed by visualization on polyacrylamide gel with Coomassie blue staining (SI Fig. 3). All recombinant repeats were first analyzed by Western blot with anti-6xHis antibody to confirm expression (Fig. 1). A single band at the expected molecular weight (
Results of indirect ELISA. Luminescent optical density values were normalized (to max 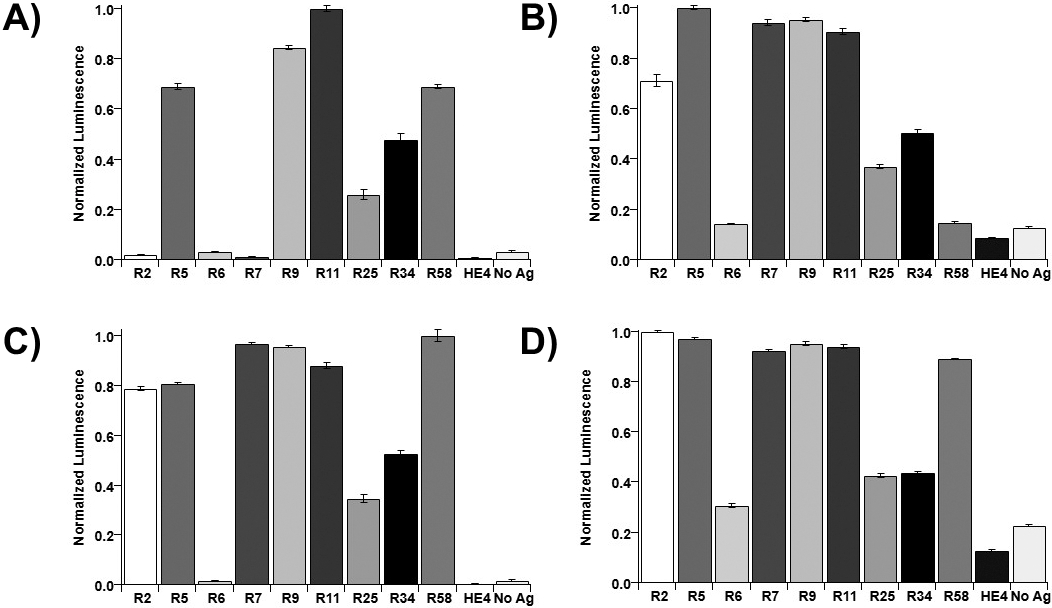
SPR data on binding of recombinant repeats from MUC16 to CA125 antibodies. Normalized (to max 
Recombinant repeats were probed via Western blot with antibodies OC125, OC125-like, M11 and M11-like, and anti-6xHis antibody as a loading control (Fig. 1). Epitope group A antibodies (OC125 and OC125-like), recognized repeats R5, R6, R9 and R25, with clear bands observed at the expected MW (
ELISA
Recombinant repeats were immobilized onto Ni-NTA-coated 96-well plates for indirect ELISA. Luminescence values were normalized to the maximum signal for each antibody (Fig. 2, shown as dot plots in SI Fig. 4). Out of the four antibodies tested, OC125 showed evidence of binding to the fewest recombinant repeats (Fig. 2A). OC125 showed maximum binding to R11 and strong binding to R5, R9, and R58, whereas R2, R6, and R7 had lower signals than the no-antigen control. OC125-like antibody (Fig. 2B) displayed strong binding to R5, R7, R9, and R11, and little binding to R6 and R58. M11 (Fig. 2C) had its maximum binding to R58, strong binding to R2, R5, R7, R9, and R11, and no apparent binding to R6. M11-like (Fig. 2D) demonstrated maximum binding to R2, with strong binding to R5, R7, R9, R11, and R58. There were no repeats that showed no binding to any of the four antibodies, but R6 had smaller luminescence signals than the other recombinant repeats. This is not conclusive evidence that binding is not occurring, as the immobilization of protein within the wells was not quantified. The slightly higher luminescence signal in no antigen control wells compared to the HE4 controls indicates that nonspecific binding of antibodies to the wells may contribute to the luminescence signal. While the dilutions of antibodies varied, and the ranges of luminescence values varied from antibody to antibody, comparing the relative affinities of each clone to the repeats with one another can give some insight into the similarity of epitopes. Each antibody showed maximum binding signal to a different tandem repeat, indicating the binding locations may vary from antibody to antibody. OC125 and the OC125-like clone do not have similar patterns of binding from repeat to repeat (Fig. 2A and B). M11 and the M11-like clone had similar binding patterns (Fig. 2C and D).
Surface plasmon resonance (SPR)
Representative sensorgrams are found in SI Fig. 6. Average corrected binding signal for each antibody interacting with recombinant repeat was normalized to the maximum signal for each antibody(Fig. 3, shown as dot plots in SI Fig. 5). A negative normalized corrected value occurs when signal resulting from non-specific binding to the sensor chip is greater than signal from recombinant repeat–antibody binding. Due to variable antibody dilution, direct comparisons based on corrected signals cannot be made. However, the pattern of normalized binding for each recombinant repeat-antibody pair demonstrates variability across combinations of recombinant repeat and antibodies. OC125 (Fig. 3A) and M11-like (Fig. 3D) show the strongest response to R58, while M11 (Fig. 3C) shows the strongest binding to R2, and OC125-like (Fig. 3B) shows the strongest binding to R5. As with the Western blot and indirect ELISA, the differences in binding patterns between the clinical antibodies and their “-like” counterparts suggest that the location of their epitopes differs. OC125 and the OC125-like antibody show variability in binding patterns: OC125 shows the strongest binding response to R58, but OC125-like shows no binding to this repeat. Differences are also seen for M11 and the M11-like antibody: M11 shows stronger binding to R2 than to R5, while M11-like has similar affinity for both repeats. R34 and R58 also show different patterns of binding for M11 and M11-like: while the two repeats show comparable binding for M11, there is stronger binding to R58 for M11-like by a factor of 7.
Heat map generated using normalized binding signals of antibodies to recombinant repeats from MUC16. Data used to generate the heat map are in SI Table 1. Any negative normalized binding signals were set to zero for the generation of the heat map.
Comparison of analytical methods
Figure 4 displays the combined data for the three assays and enables visualization of differences in binding across recombinant repeat/antibody combinations (the raw data used to generate the heat map are found in SI Table 1). We observe that the extent of binding differs across the three analysis methods. One potential explanation of these differences is the nature of the assays: prior to Western blotting, recombinant repeats were denatured and treated to reduce disulfide bonds, while in ELISA and SPR assays the recombinant repeats remained as expressed with disulfide bonds intact. While it is not certain that the folded state of the expressed and purified recombinant repeats is structurally identical to the native state of a MUC16 tandem repeat in vivo, the discrepancy in antibody binding ability when recombinant protein is denatured supports the hypothesis that the epitopes for these antibodies is conformational [29, 36]. R6 and R25 are notable exceptions to the pattern. For these two recombinant repeats, Western blot results indicate antibody-repeat binding and the other binding analysis methods showed little evidence of interaction. In SPR experiments, R6 and R25 consistently had significantly lower immobilization signal response than other repeats, and yields during NTA affinity chromatography-based purification were consistently lower for these two recombinant proteins compared to the other seven. This observation implies that the lower binding signals in SPR and ELISA for R6 and R25 may result at least in part from inefficient immobilization of these recombinant repeats. The patterns of which repeat-antibody combinations show strong, weak, or minimal binding evidence look similar between ELISA and SPR, two methods in which the recombinant repeats were not denatured and were immobilized in the same orientation. ELISA generally showed more consistent binding signals across recombinant repeats than did SPR. Corrections for nonspecific binding could account for these differences. ELISA results may have been affected by variable immobilization or nonspecific binding, which were not tracked through data collection. In SPR assays nonspecific binding signal was subtracted from binding signal.
Working hypothesis regarding the nature of the CA125 epitopes
Structural models for each of the nine tandem repeat proteins were obtained using the iTasser server [41, 42, 43]. In all cases, the top threading template was 7sa9, the human MUC16 SEA5 domain reported by White et al. [23]. This structure was also the top identified structural analog in the Protein Data Bank for all nine tandem repeat proteins, with good topological similarity, inciated by TM-scores of 0.749, 0.760, 0.760, 0.761, 0.763, 0.761, 0.758, 0.765, and 0.758 for repeats R2, R5, R6, R7, R9, R11, R25, R34, and R58, respectively, where a TM score of 1 indicates a perfect match. The sequence identity of the tandem repeats with 7sa9 were 0.861, 0.967, 0.943, 0.885, 0.795, 0.811, 0.992, 0.811, and 0.836, indicating that this previously reported model is an excellent template for other MUC16 tandem repeat domains. The predicted models overlay the MUC16 SEA5 domains well (SI Fig. 7), with the notable exception of the proline-serine-threonine (PST) rich region which is largely unstructured in the iTasser predictions of the tandem repeats. The SEA5 domain structure does not contain the PST region. The two major accessible faces noted by White et al. in their analysis of the crystal structure of SEA5 are seen in the tandem repeat domain proteins modeled here. It is not likely that the differences in antibody binding that we observe results from structural differences at these faces. Structural divergence between the SEA5 model (which contains a Serine at position 66) and the subset of tandem repeat proteins (R2, R7, R9, R11, R34, and R58) that contain Proline at that position is notable in the unstructured loop between beta strands that comprise the “C-loop” [32]. The center of the C-loop contains highly charged and polar amino acids and is found, in the model predicted by i-Tasser, to be adjacent to another loop that could be close enough to comprise a conformational epitope. Based on these studies, we hypothesize that the CA125 epitope involves the C-loop and charged and polar amino acids in the neighboring coil (residues 82–89). Notably, the tandem repeat protein that was found to be least immunogenic by Western blotting (R7) has aspartic acid replaced by asparagine within the loop that may be involved in conformational epitope presentation. Determining the exact location of the epitopes is the focus of our ongoing work.
Conclusions
This study contributes to ongoing efforts to identify the peptide epitopes of CA125. We anticipate that CA125 can be a more informative clinical biomarker if the nature of its recognition by antibodies is elucidated. Such knowledge may enable development of affinity reagents that recognize all repeats of MUC16. The current clinical test may under-report concentrations of CA125 in blood samples if the antibodies used for detection do not reliably recognize the subdomains in the immunogenic region, as data reported here suggest. Detection of smaller amounts of CA125 may be possible, enabling earlier detection of CA125 resurgence, which correlates strongly with cancer recurrence. Future work following from this study includes structural characterization of free and antibody-bound recombinant repeats and the use of recombinant repeats as targets for the generation of novel affinity reagents such as nucleic acid aptamers, immunoaffinity reagents, and vaccines.
Author contributions
Conception: CWW, EKH, LM, RJW.
Interpretation or analysis of data: CWW, EKH, LM, RJW.
Preparation of the manuscript: CWW, EKH, LM, RJW.
Revision for important intellectual content: CWW, EKH, LM, RJW.
Supervision: RJW.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-220191.
sj-docx-1-cbm-10.3233_CBM-220191.docx - Supplemental material
Supplemental material, sj-docx-1-cbm-10.3233_CBM-220191.docx
Footnotes
Acknowledgments
The authors thank Bill Boggess and the Notre Dame Mass Spectrometry and Proteomics Facility for technical assistance and Professor Brian Blagg for use of a ChemiDoc imager. The authors thank Professor Matthew Champion for helpful discussions and access to equipment for bacteria culture. Naviya Schuster-Little provided the structural alignment in SI ![]() , and we thank her for many helpful discussions about protein structure prediction and comparisons. Daryl Good and Marko Jovic at Nicoya were pivotal in developing and optimizing SPR assays. This work was supported by award R21CA267532 from the National Cancer Institute and a Medical Research Program award from Tell Every Amazing Lady
, and we thank her for many helpful discussions about protein structure prediction and comparisons. Daryl Good and Marko Jovic at Nicoya were pivotal in developing and optimizing SPR assays. This work was supported by award R21CA267532 from the National Cancer Institute and a Medical Research Program award from Tell Every Amazing Lady