Protein kinase R (PKR) can suppress various types of solid tumors by inducing cellular oxidative stress and apoptosis. Likewise, Slaidorside, a plant flavonoid, was shown to have anti-tumorigenesis in many solid tumors.
OBJECTIVE:
This study evaluated anti-tumorigenesis of Salidroside in HT29 colorectal cancer and investigated if the underlying mechanism involves activation of PKR.
METHODS:
Control or PKR deficient cells were cultured in DMEM media treated with 100 M Salidroside and cell survival, apoptosis, and other biochemical-related markers were evaluated.
RESULTS:
Salidroside significantly reduced cell survival and proliferation and increased the release of lactate dehydrogenase (LDH) and levels of single-stranded DNA (ssDNA). It also increased the protein levels of caspases 3 and 8. Concomitantly, Salidroside increased the protein level and activity of PKR and increased the expression of its downstream targets, p-eIF2 (Ser), p53 MAPK, and p53. On the contrary, it inhibited the nuclear activation of STAT-3 and NF-B p65. In PKR deficient cells, the partial effects of Salidroside on cell survival, proliferation, and apoptotic markers were observed coincided with no effects on the expression of eIF-2, and JNK, p53, p38 MAPK, and caspase 8 but with a significant decrease in the nuclear activities of STAT3 and NF-B.
CONCLUSION:
Salidroside suppresses the tumorigenesis of HT29 CRC by increasing activation of eIF-2 and JNK and upregulation of p53, p38 MAPK, and caspase-8 through upregulating and activation of PKR. However, the tumor suppressor effect of Salidroside requires also inhibition of STAT3 and NF-B in a PKR-independent mechanism.
Colorectal cancer (CRC) is one of the most lethal cancers in old and young males and females worldwide ( 30% in both sexes) with a projection to increase by 60% in 2030 [1]. Risk factors for CRC include a high meat diet and smoking [1, 2]. Reactive oxygen species (ROS) and inflammation remain the major well-known tumorigenic pathways in CRC [2, 3]. Unfortunately, CRC still a very complicated disorder that requires more exploration and specific drug development.
Currently, several lines of evidence have pointed out the emerging role of the protein kinase R (PKR) in the pathogenesis of numerous types of solid tumors and other chronic inflammatory disorders and metabolic disorders, as well as, neurodegenerative disorders [4, 5, 6]. In general, PKR is a 551 amino acid long serine-threonine kinase that is encoded in humans by the EIF2AK2 gene [4, 6]. It is mainly induced by double-stranded RNA (dsRNA) and interferon- (INF-) during the viral pathogen invasion and plays a significant role as an antiviral agent through suppression of mRNA transcription, protein synthesis, and viral replication, as well as induction of cell apoptosis [5, 7, 8]. However, the upregulation and activation of PKR can also be induced in other non-viral mechanisms, including inflammatory cytokines, energy excess, over-nutrition, lipid accumulation, calcium stress, irradiation, reactive oxygen species and oxidative stress, endoplasmic reticulum (ER stress), Lipo-stress, amyloid- (A) peptide accumulation, and many drugs [4, 5].
However, the contribution of PKR in tumorigenesis of different types of solid tumors was shown to be a dual effect of either promoting or suppressing tumor progression. Besides, PKR levels and activity were significantly increased or decreased in Serval types of tumors [5, 9]. Indeed, reduced levels and activation of PKR were shown in head, skin, lung, and colon cancer where its activation inhibited tumor progression and improved the prognosis, thus suggesting a tumor suppressor effect of this enzyme [10, 11, 12, 13]. On the other hand, another line of evidence has shown overexpression of PKR in thyroid, colon, breast, liver, lung, and skin cancers, as well as in acute myeloid leukaemia, thus challenging the above-mentioned studies that PKR is a tumor suppressor [9, 14, 15, 16, 17, 18]. Such duality of effects was shown due to different experimental settings (in vivo vs in vitro) and tumor type and stage and is dependent on other activated signaling pathways, thus leaving a debate on the overall effect of PKR function in cancer [5].
However, the tumor suppressor effect of PKR is believed to be due to its ability to induce cell apoptosis and arresting the cell cycle through its wider effect on various signalling pathways that control these pathways. Within this view, it was shown that PKR induces cell apoptosis by phosphorylation and activation and of the Eukaryotic Initiation Factor 2 (eIF-2), thus impairing its activity which ultimately leads to inhibition of protein synthesis [4, 5]. In addition, PKR can induce cell apoptosis and inhibit cancer cell invasiveness in an EIF-2-independent manner and through different mechanisms, including increasing the transcription and translation of Fas and apoptotic Bcl-2 effector proteins, activation of caspases 3/8/9 [19, 20]. Furthermore, PKR induces cell apoptosis through interaction with P53 and inhibits cell invasiveness through upregulation of ATF-3 transcription factor [5, 9]. Besides, PKR is a major stimulator of the apoptotic signalling pathways, including kinase (JNK) and p38 mitogen-activated protein kinase [21, 22]. On the contrary, PKR has been shown to activate the transcription factor nuclear factor-kappa beta (NF-B) which plays a significant role in promoting tumor proliferation, growth, stemness, and invasion [5, 19].
Nonetheless, the plant Kingdome remains a rich source of anticancer-derived drugs, some of which having minimal side effects rather than synthetic drugs [23]. Salidroside is a major flavonoids glycoside isolated from Rhodiola rosea (R. rosea L) [24]. Covering data are showing a potent tumor-suppressive effect of Slaidorside against a variety of solid tumors, including thyroid, renal, bladder, breast, lung, CRC, and colon cancers [25, 26, 27]. Among all these studies, the confirmed mechanisms of protection involved inhibition of ROS generation, expression of BCl2 and signalling through PI3K/Akt/mTOR, as well as activation of caspase-3 dependent cell apoptosis, and inhibition of JAK2/STAT3.
However, the effect of Salidroside on the expression and activation of PKR was never investigated either in normal or cancer cells. Therefore, in this study, we aimed to investigate if the tumor suppressor effect of Salidroside is mediated, at least, by modulating the expression, activation, and downstream signalling of PKR in the HT29 CRC cell line. Also, the expression of serval PRK downstream targets including JNK, P38 MAPK, NF-B, ATF-3, STAT1/STAT3 were targeted.
Materials and methods
Cell culture and treatments
The Human CRC cell line, HT29 was purchased from the American Type Culture (USA) and were cultured (at 85% confluent) in DMEM supplied with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin, and 0.1% streptomycin (ThermoFisher Scientific) (5% CO2, 37C) (He et al., 2017). They were seeded in and incubated under similar conditions for 24 h.
Small interfering RNAs (siRNAs) and cell treatment
PKR siRNA (h) (Cat. No. sc-36263), Control siRNAs (Cat. No. sc-37007), siRNA Transfection Reagent (Cat. No. sc-29528), siRNA Transfection Medium (Cat. No. sc-36868) and siRNA Dilution Buffer (Cat. No. sc-29527) were purchased from Santa Cruz Biotechnology, TX, USA. Transfection reagent was performed according to the manufacturer’s instructions. In brief, cells were initially seeded at a density of seed 2 10 cells per well in a 2 ml antibiotic-free normal growth medium supplemented with FB in O2 incubator at 37C in a CO incubator to reach a confluence of 60–80%. The siRNA of PKR (6 l of 10 M) or control siRNA (8 l of 10 M) as well as the transfection reagent (6 l) were diluted individually with 100 l transfection media and then mixed and incubated for 30 min to prepare the transfection reagent working mixture. The transfection working mixture was added to 0.8 ml of the transfection media (free of serum and antibiotic) and the whole 1 ml mixture was overlaid onto the washed cells and incubated for 5 h. after that, 1 ml of 2x growth media was added to each transfection and incubated for 18 h which was then removed and replaced with 1x growth media and further incubated for 48 h. To confirmed the results, protein expression of PKR was confirmed by western blotting.
Cell treatment
Depending on the experimental procedures, cell (HT29 or knocked-down HT29 cells) were cultured at a density of 2 10 (5% CO, 37C) in 96 well plates in the growth culture media and treated with Salidroside (Cat. No. SMB00072, Sigma Aldrich, St Louis, MO, USA) at a final concentration of 100 M. This concentration was based on the initial dose-response curve of the effect of increasing doses (0–200 M) of Salidroside on cell survival, proliferation, levels of lactate dehydrogenase (LDH), and cell apoptosis.
Determination of cell viability
The rate of cell viability was measured using a cell viability counting kit-8 (CCK-8) (Cat No. CK04-13, Dojindo, Japan) in accordance with the manufacturer’s instruction. In brief, 100 l of the growth media was discarded and replaced with a new fresh medium containing 10% of working CCK8 solution, incubated for 3 h at 37C and reading the absorbance (ABS) at 450 nm (Spectramax plate reader; Model M2, Molecular Devices, USA).
Measurements of LDH activity
All treated cells were centrifuged (200xg, 5 min, 4C) to collect the supernatants. LDH activity in all collected supernatant was measured using a commercial kit (Cat. No. ab102526; Abcam, Cambridge, UK). In brief, the test relies on the conversion of NAD in the presence of LDH to NADPH which can be measured and detected using a special probe. In the test, 2 l LDH substrate buffer was mixed with 48 l of LDH assay buffer to generate a reaction mixture that was added to each sample or standard (20 l). The measurements of the ABS were performed at different time intervals at 450 nm (Spectramax plate reader; Model M2, Molecular Devices, USA) and LDH levels were obtained from the standard curve.
Measurement of cell proliferation
Cell proliferation was determined by Bromo-2-deoxyuridine (BrdU) ALISA kit (Cat. No. 11647229001; Roche Diagnostics, Indianapolis, IN, USA). The principle of the test relies on the incorporation of BrdU into newly synthesized cellular DNA, which can be then detected by using specific BrdU-peroxidase (POD) and a substrate reagent, 3, 3, 5, 5-tetramethylbenzidine. Briefly, the cells were cultured with various treatments in a final volume of 100 l for 24 h. then10 l BrdU was added to each well and incubated for 2 h at 37C. Then, the media were removed, and 200 l FixDena was added to each well and incubated for 30 min at 23C. The FixDena solution was removed and 100 l POD solution was added to each well and incubated at 23C for 90 min. The solution was then removed and each well was washed three times with 200 l of the washing solution (PBS). Then 10 l of substrate reagent was added to each well and incubated at 23C for 15 min until the color was developed. Absorbance was read at 370 nm with a reference range at 492 nm (Spectramax plate reader, model M2, Molecular Devices, USA). Cell proliferation was calculated as the percent of controls.
Determination of apoptosis (ssDNA assay)
The levels of ssDNA in all tested samples were measured using an ELISA kit (Cat No. APT225, Millipore, USA) per the manufacturer’s instruction. Briefly, the cells were centrifuged (200xg, 5 min, 4C) to collect pellets. Pellets were then fixed with 200 l of the supplied fixative reagent and kept at 23C for 30 min, after which the fixative was discarded and all plates were dried at 23C for 2 h. After that, 100 l of formamide solution was added to each well, incubated the plate was incubated at 23C for 10 min, and then heated at 75C for 10 min to denature DNA. Then, all plates were cooled at 4C and the formamide was removed. Next, 3% 100 l skim milk (prepared in distilled water) was added to all wells and incubated for 1 h at 37C. Then, the milk was removed and cells of all wells were incubated with 100 l mouse monoclonal anti-ssDNA antibody (supplied with the kit) for 30 min at 23C, then washed with 250 l of 1x of the provided washing buffer (3 times). Then, 200 l of the provided HRP-secondary anti-mouse secondary antibody was added incubated for 30 min at 23C. After that, 100 l ABTS solution was added to all wells and incubated for another for 30 min at 23C. to allow sufficient binding of the HRP. The reaction was terminated by the addition of 100 l stop solution and the ABS was read at 405 nm (Spectramax plate reader, Model M2, molecular devices, USA).
Migration and invasion assays
The migration and invasion rates of both cell lines were, according to the method previously described in our laboratories [28]. For cell migration, the cells (5 10 cells, 100 l) were plated on the top of the Transwell chambers (8-m pores and a membrane diameter of 6.5 mm) (Corning Costar, NY, USA) and treated as mentioned whereas the lower chamber was filled with 500 l 10%. The whole setting was incubated for 24 h. Then, the cells in the upper chamber were wiped and those in the lower chamber were fixed with methanol for 10 min at 23C and then stained with 0.2% crystal violet for another 10 min at 25C. Stained cells were counted using a light microscope in 4 random fields and average readings were presented. Cell invasion was performed in the same way, but using Corning Transwell chambers of 5-mm pore size. Before the test, the upper chamber was first coated with 50 l Matrigel (Cat. No. DLW354263, Sigma Aldrich, UK). During the invasion test, cells were first starved in a serum-free medium for 3 h and then loaded onto the upper chamber.
Western blots
Cell pellets of all treatments were homogenized in 250 l RIPA buffer lysis kit (Cat. No. 89900, ThermoFisher) supplied with 5 l protease inhibitor cocktail (Cat. No. P8340 Sigma-Aldrich, MO, USA). The samples were centrifuged 10000xg for 10 min to collect the supernatant which was collected and stored at 80C until use. Protein levels in all fractions were determined by a Bradford assay using a commercial kit (Cat. No 23300, ThemoFisher Scientific, MA, USA). The Western blot procedure was performed as previously described in our labs [29]. Proteins from all groups (40 g/well) were separated by SDS Page and transferred onto nitrocellulose membranes. Membranes were first incubated, for 2h at room temperature, with rotation, with the primary antibodies against PKR (Cat. No. 3072, 1:1000, 74 kDa), p-PKR (Thr) (Cat. No. 3075, 1:1000, 74 kDa), eIF-2 (Cat. No. 5324, 1:1000, 38 kDa), p-eIF-2 (Ser) (Cat. No. 5324, 1:1000, 38 kDa), JNK (Cat. No. 9252, 1:1000, 46/54 kDa), p-JNK (Thr/Tyr) (Cat. No. 9255, 1:1000, 46/54 kDa), p38 MAPK (Cat. No. 8690, 1:1000, 40 kDa), p53 (Cat. No. 2527, 1:1000, 53 kDa), STAT3 (Cat. No. 9139, 1:1000, 86 kDa), p-STAT3 (Tyr) (Cat. No. 9145, 1:1000, 53 kDa), NF-k p65 (Cat. No. 8242, 1:1000, 65kDa), p-NF-B p65 (Ser) (Cat. No. 3033, 1:1000, 53 kDa), cleaved caspase-3 (Cat. No. 9661, 17/19 kDa, 1:500), caspaspe-8 (Cat. No. 9746, 1:1000, 57 kDa), -actin (Cat. No. 3700, 45 kDa, 1:2000), lamin B1 (Cat. No. 12255, 1:1000, 68 kDa) (Cell signalling Technology, MA, USA). Next, membranes were incubated with appropriate corresponding horseradish peroxidase (HRP)-conjugated secondary antibody for 2 h at room temperature with rotation. All antibodies were prepared in TBST buffer and all membranes. Membranes were stripped with a commercially available stripping buffer (Cat. No. 21059, ThermoFisher Scientific) up to 5 times and the detection of the phosphorylated forms was done first, and the -actin the last. A control known sample was run on all gels for normalization of the target protein. Band intensities were detected using a Pierce ECL kit (ThermoFisher, USA, Piscataway, NJ), scanned using a C-Di Git blot scanner (LI-COR, NE, USA), and analyzed using the scanner associated software.
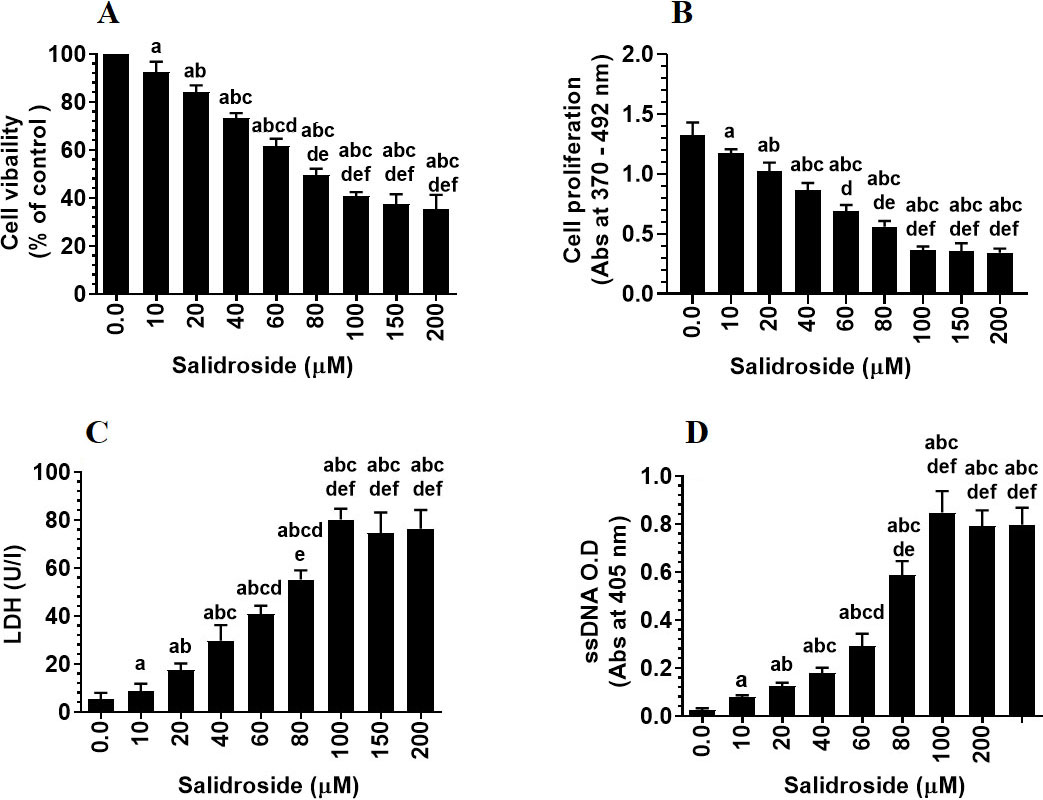
Salidroside induces a dose-response, decrease in cell survival (A) and cell proliferation (B), and a dose-response increase in LDH releases (C) and content of single-stranded DNA (ssDNA) (D) in HT29 colorectal cell line. HT29 cells were cultured in DMEM for 24 h under 5% CO and 37C and treated with increasing concentrations of Salidroside (0–200 M). Data are presented as mean SD of three experiments/treatment each performed in triplicate. a: vs. control (0.0 M), b: vs. 10 M, c: vs. 20 M, d: vs. 40 M, e: vs. 60 M, and f: vs. 80 M.
Statistical analysis
GraphPad Prism statistical software package (V8) was used for the analysis of data. Data were analyzed by one-way ANOVA, followed by Duncan’s Multiple Range Test. The significance among parameters was determined at 0.05. Data are presented as mean SD.
Results
Salidroside inhibits HT29 survival and invasiveness and stimulates cell death, at least by activation of PKR/eIF-2
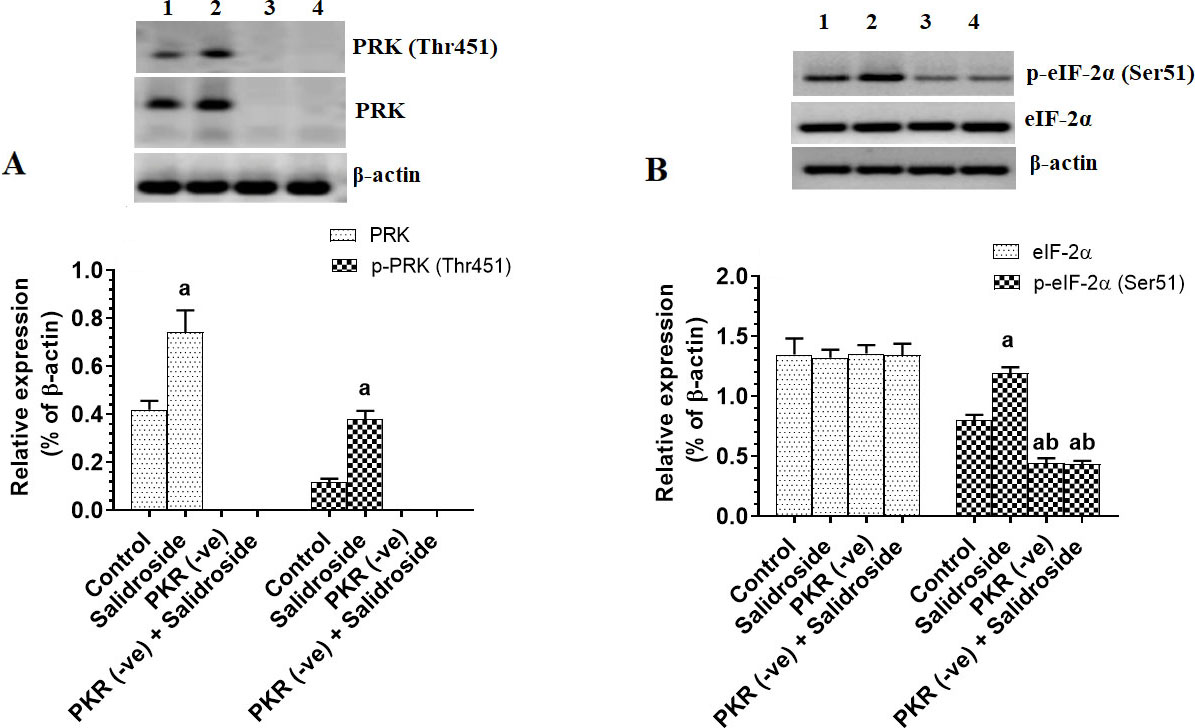
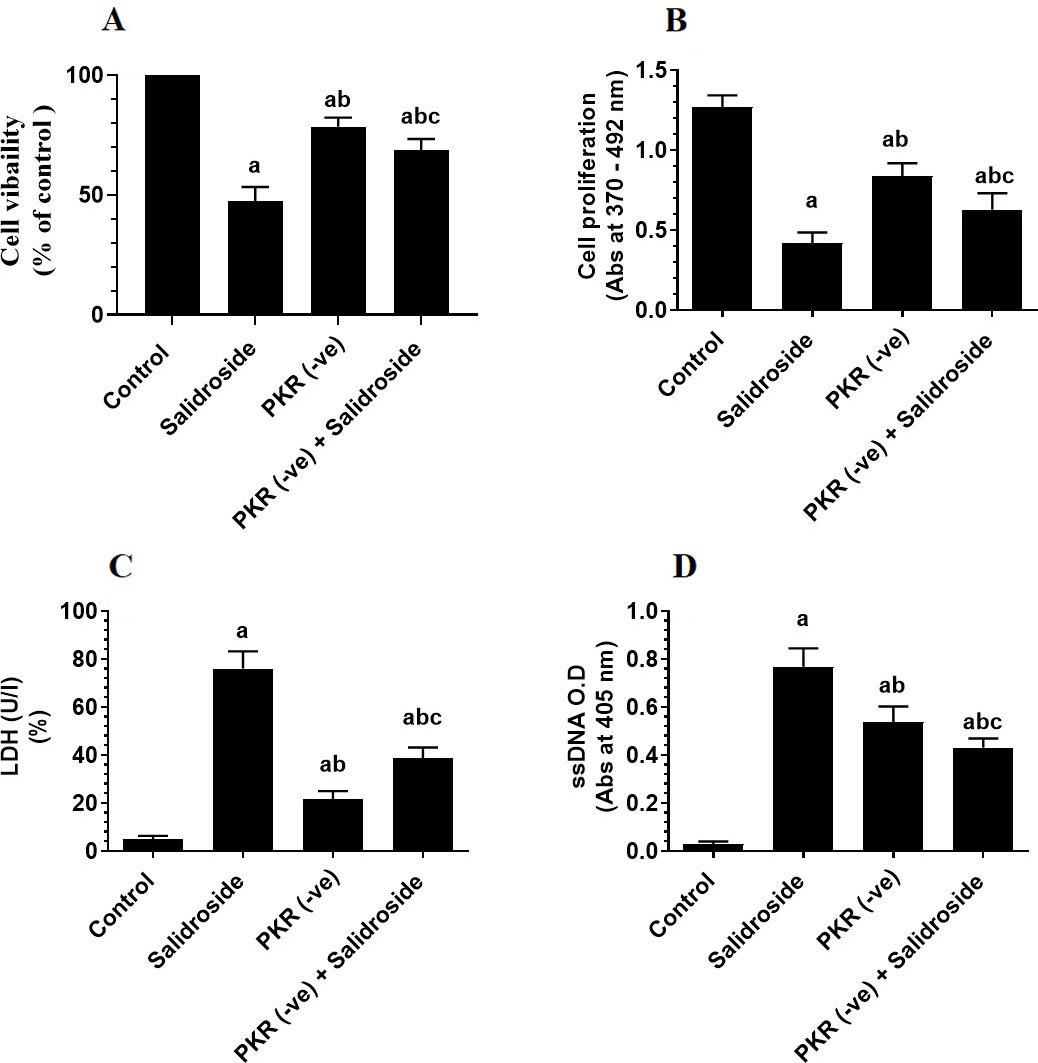
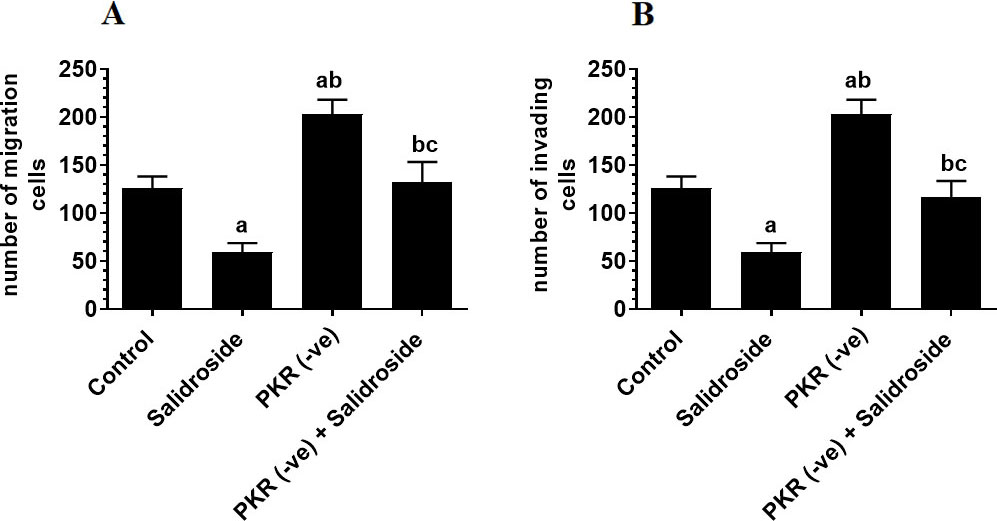
To test the tumor suppressor effect of Salidroside on the HT29 CRC cell line, we have treated the cells with increasing concentrations of the drug. Salidroside, in a dose-dependent manner, decreased cell survival and proliferation and increased the release of LDH and levels of ssDNA, a marker of cell apoptosis (Fig. 1A–D). The maximum effect of Salidroside on these parameters was shown at concentrations of 100, 150, and 200 M where no significant variation was seen between them (Fig. 1A–D). When tested at a dose of 100 M, Salidroside significantly increased the protein levels of PKR, p-PKR (Thr), and p-eIF-2 (Ser) as compared to control cells, thus indicating upregulation of PKR and activation of both PKR and eIF-2 (Fig. 2A and B). Concomitantly, Salidroside at a concentration of 100 M significantly reduced cell survival and proliferation (Fig. 3A and B), increased LDH release and ssDNA levels (Fig. 3C and D), and suppressed cell migration and invasion as compared to control cell (Fig. 4A and B). These data confirm the tumor suppressor effect of Salidroside and highlight the involvement of activation of PKR/eIF-2 in this process.
Salidroside increases protein level and activity of protein kinase R (PKR) and subsequently activates the Eukaryotic Initiation Factor 2 (eIF-2) in HT29 colorectal cells. HT29 control or PKR deficient cells were cultured in DMEM for 24 h under 5% CO and 37C and treated with Salidroside (100 M). Data are presented as mean SD of three experiments/treatment each performed in triplicate. a: vs. control (untreated control cells) (Lane 1), b: vs. Salidroside-treated control cells (Lane 2), c: vs. PKR-deficient cells (Lane 3). Lane 4 represents a sample that was taken from PKR-deficient cells Salidroside.
Deletion of protein kinase R (PKR) partially ameliorates the inhibitory effect of Salidroside on cell survival (A) and proliferation (B) and its stimulatory effect on LDH release (C) and the increase in levels of single-stranded DNA (ssDNA) (D) in HT29 colorectal cell line. HT29 control or PKR deficient cells were cultured in DMEM for 24 h under 5% CO and 37C and treated with Salidroside (100 M). Data are presented as mean SD of three experiments/treatment each performed in triplicate. a: vs. control (untreated control cells), b: vs. Salidroside-treated control cells, c: vs. PKR-deficient cells.
On the other hand, the deletion of PKR significantly reduced the eIF-2 levels (Fig. 2B), partially reduced cell survival and proliferation (Fig. 3A and B), and partially increased levels of LDH and ssDNA (Fig. 3C and D), as compared to control cells. Also, it significantly increased cell migration and invasion as compared to control cells (Fig. 4A and B), thus indicating that basal levels of PKR are crucial for tumorigenesis of HT29 cells.
Deletion of PKR increases cell invasion (A) and migration (B) whereas treatment of Salidroside inhibits these events in both control and protein kinase R-deficient HT29 colorectal cells. HT29 control or PKR deficient cells were cultured in DMEM for 24 h under 5% CO and 37C and treated with Salidroside (100 M). Data are presented as mean SD of three experiments/treatment each performed in triplicate. a: vs. control (untreated control cells), b: vs. Salidroside-treated control cells, c: vs. PKR-deficient cells.
However, when Salidroside was incubated with PKR-deficient HT29 cells, it failed to alter the expression of eIF-2, partially decreased cell survival and proliferation, partially increased levels of LDH and ssDNA, and partially suppressed cell migration and invasion as compared to control Salidroside-treated cells (Fig. 2B, Fig. 3A–D, and Fig. 4A and B). Although basal levels of PKR are shown to promote CRC progression in HT29 cells, these data suggest that activation of PKR by Salidroside is a tumor suppressor event but not the only signaling pathway induced but Salidroside.
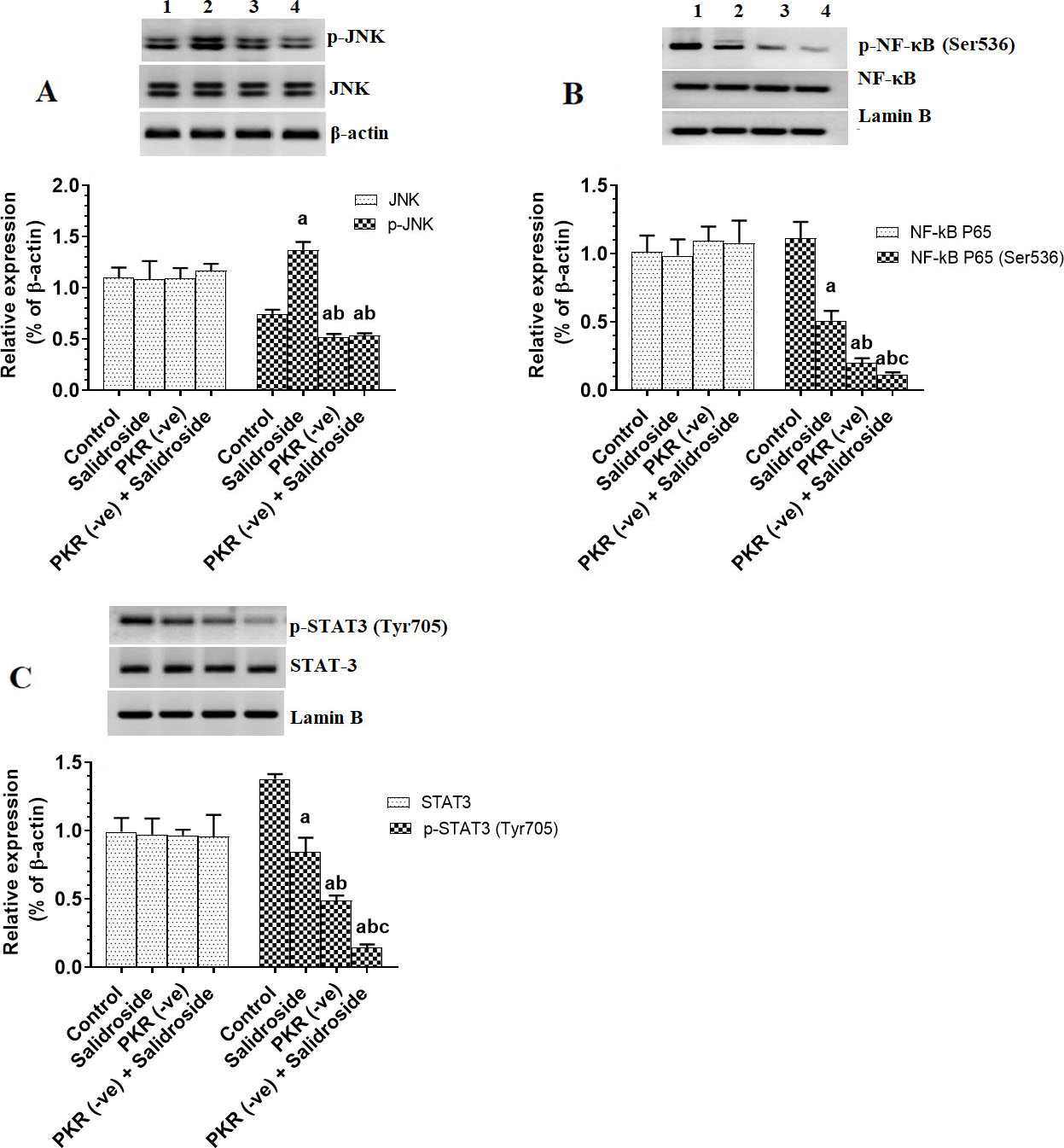
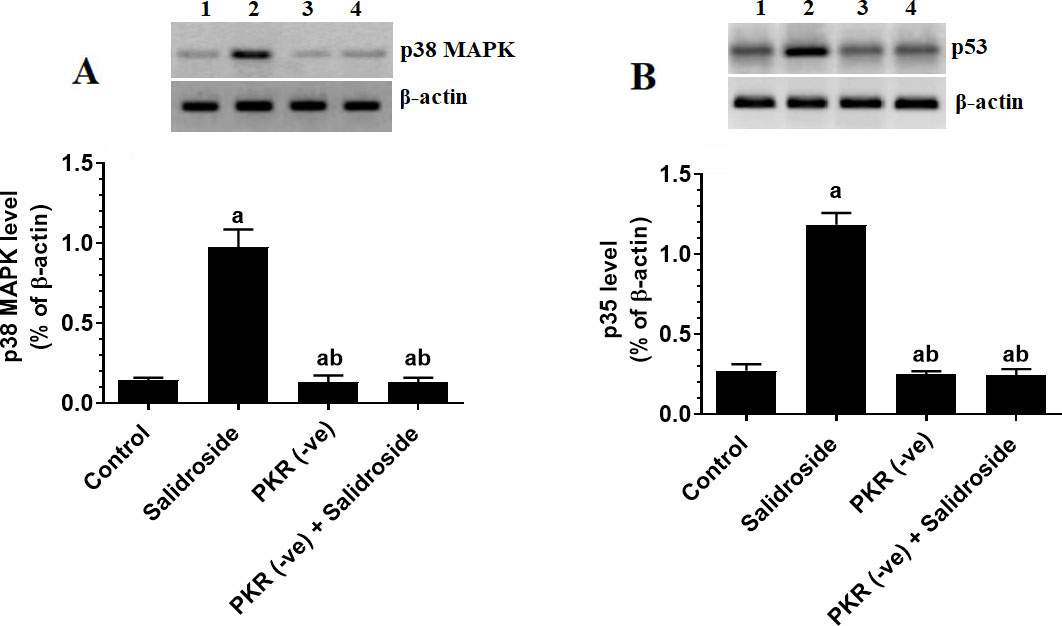
Salidroside activates JNK and upregulated p53 and p38 MAPK only in control cells, but inhibits NF-B and STAT3 in both control and PKR-deficient cells
JNK, p38, p53, NK-B, and STAT-3 are major targets of PKR (Garcia et al., 2017). Indeed, with no alteration in total levels of JNK and nuclear levels of NF-B and STAT3 deletion of PKR in HT29 cells significantly reduced the total protein levels of p-JNK and nuclear levels of p-NF-B (Ser) and p-STAT3 (Ty) (Fig. 5A–C) with a parallel decrease in total protein levels of p38 MAPK and p53 (Fig. 6A and B). On the other hand, treating the HT29 cells with 100 M Salidroside for 24 h significantly increased levels of p-JNK, p53, p38 MAPK but significantly lowered nuclear levels of p-NF-B (Ser) and p-STAT3 (Ty) (Fig. 5A–C and Fig. 6A and B). Interestingly, although Salidroside was unable to alter protein levels of p-JNK, p53, and p38 MAPK in PKR-deficient cells as compared to PKR-deficient cells, levels of p-NF-B (Ser) and p-STAT3 (Ty) showed the maximum decrease in the PKR-deficient cells Salidroside (Fig. 5A–C and Fig. 6A and B). These data suggest that Salidroside induced activation of JNK, p38, and MAPK are PKR-dependent. Besides, it can be concluded that Salidroside is a potent inhibitor of NF-B and STAT3.
Salidroside increases the activation of JNK only in control HT29 colorectal cells, but inhibits the nuclear activity of NF-B and STAT3 in both control and protein kinase R (PKR) -deficient cells. HT29 control or PKR-deficient cells were cultured in DMEM for 24 h under 5% CO and 37C and treated with Salidroside (100 M). Data are presented as mean SD of three experiments/treatment each performed in triplicate. a: vs. control (untreated control cells) (Lane 1), b: vs. Salidroside-treated control cells (Lane 2), c: vs. PKR-deficient cells (Lane 3). Lane 4 represents a sample that was taken from PKR-deficient cells Salidroside.
Salidroside upregulated p38 MAPK and p53 only in control HT29 colorectal cells. HT29 control or PKR-deficient cells were cultured in DMEM for 24 h under 5% CO and 37C and treated with Salidroside (100 M). Data are presented as mean SD of three experiments/treatment each performed in triplicate. a: vs. control (untreated control cells) (Lane 1), b: vs. Salidroside-treated control cells (Lane 2), c: vs. PKR-deficient cells (Lane 3). Lane 4 represents a sample that was taken from PKR-deficient cells Salidroside.
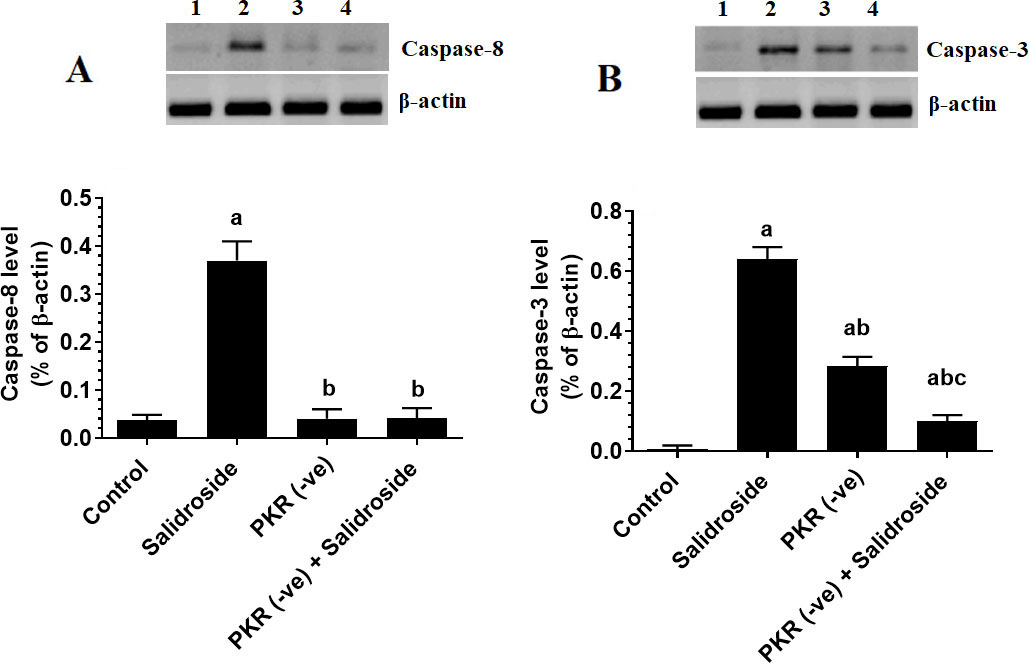
Salidroside upregulates caspase-8 only in control cells but upregulates caspase-3 in both control and PKR-deficient cells
Treating the HT29 cells with Salidroside significantly upregulated caspase-3 and caspase-8 as compared to control cells (Fig. 7A and B). However, the deletion of PKR did not affect the protein levels of caspase-8 but partially reduced protein levels of caspase-3 as compared to control cells (Fig. 7A and B). Notably, treating the PKR deficient cells with Salidroside didn’t affect protein levels, but further reduced the expression of caspase-8 as compared to PKR-deficient cells (Fig. 7A and B). These data suggest that the upregulation of caspase-8 by Salidroside is PKR-dependent, whereas the upregulation of caspase-3 involves at least PKR but requires activation of other pathways.
Salidroside downregulates casapase8 only in control HT29 colorectal cells, but downregulates caspase-3 in both control protein kinase R (PKR)-deficient cells. HT29 control or PKR-deficient cells were cultured in DMEM for 24 h under 5% CO and 37C and treated with Salidroside (100 M). Data are presented as mean SD of three experiments/treatment each performed in triplicate. a: vs. control (untreated control cells), b: vs. Salidroside-treated control cells, c: vs. PKR-deficient cells.
Discussion
In this study, we have confirmed the tumor suppressor effect of Salidroside in an HT29 CRC cell line and have shown that it is not only able to induce cell apoptosis and inhibit proliferation but also suppress their invasion and migration. However, the novelist finding of this study is that we are showing a collaborative mechanism by which Salidroside exerts its anti-tumorigenesis effects which involve 1) upregulation and activation of PKR and subsequent upregulation of p38 MAPK and p53 and activation of eIF-2 and JNK activation and 2) PKR independent mechanisms which involve inhibition of NF-B and STAT3 activation.
Initially, Salidroside, and in a dose-dependent manner inhibited HT29 cell proliferation and survival and significantly stimulated their death. These data were also confirmed by a sustained increase in cleaved caspase-3, which is considered a classic marker of cell death. Such findings remain confirmatory to many other previous studies that have shown a tumor suppressor effect of Salidroside against many solid tumors including the CRC. Indeed, Salidroside inhibited cell growth, proliferation, and invasion of MG63 and U2OS human osteosarcoma, WRO differentiated thyroid, and SW1116 colon cancer cell lines through inhibition of the JAK2/STAT-3 signalling pathway [27, 30, 31]. Also, Salidroside inhibited the growth, proliferation, and invasion of A549 lung cancer cells and suppressed metastasis in breast cancer cells by inhibiting ROS generation and activation of p38 MAPK [32]. In HCT-116 CRC cancer cells, Salidroside inhibited cell survival and induced cell death by inhibiting different targets, including mTOR, STAT3, and NF-B. However, in HT29 CRC, Salidroside tumor-suppressive effect was mediated by inhibition of the PI3K/Akt/mTOR signaling axis [33].
On the other hand, a unique finding in this study is that we also report a new protein target of Salidroside in HT29 CRC. Herein, we are providing the first evidence in the literature that treatment with Salidroside may affect cell function by inducing an upregulation and activation of PKR. We have specifically targeted this protein due to its multifunction on different intracellular cell signalling and its pre-established role in cancer initiation and progression [6, 8]. In general, PKR is an interferon (IFN) -inducible double-stranded RNA (dsRNA) protein kinase that normally responds to viral infection by inhibiting transcription, translation, and proliferation, thus inducing cell apoptosis [6]. However, the activation of PKR requires several conformational changes in which homodimerization was shown to be mandatory for PKR activation. Also, autophosphorylation at sites Thr and Thr are indispensable for PKR homodimerization and increased its catalytic activity [7, 8]. This has been also shown in this study where Salidroside didn’t only increase the protein levels of PKR but also increased its phosphorylation at Thr, thus activation.
Under normal conditions, the cytoplasmic levels of PKR in normal cells are kept at low levels [6]. However, under a viral infection or other stress conditions such as ROS, UV radiation, ER stress, inflammatory cytokines, etc., cytoplasmic levels and activity of PKR are rapidly increased [5, 6]. In cancer cells, levels of PKR were either increased or decreased and had contradictory effects of either promoting or inhibiting cell survival [5, 9]. Indeed, reduced levels of PKR were shown on the neck, head, melanoma, lung, and colon cancers and were positively correlated with the prognosis of the disease [10, 13]. As an apoptotic kinase, PKR can induce cell apoptosis by phosphorylation and activation of eIF-2 (at Ser) and by direct activation of the FADD/caspase-8/caspase- 3 and caspase-9/APAF pathways [13, 20]. However, the tumorigenic role of PKR is mediated by activation of STAT-3 and NK-B [4, 5].
Associated with the increase in the level and activity of PKR and cell death in Salidroside-treated control HT29 cells, we have also found a significant increase in phosphorylation of eIF-2 at Ser and higher protein levels of caspase-3 and caspase-8 in these cells. These data suggest a possible role of PKR in regulating the eIF-2 pathway and acting as a tumor suppressor. To confirm this, we have silenced PKR in these cells and then treated them with the highest effective dose of Salidroside. Interestingly, this resulted in partial effects of Salidroside on cell survival, LDH release, and cell death. Also, Salidroside failed to increase levels of elF-2 in PKR-deficient cells, partially increased protein levels of caspase-3, and partially inhibited cell migration and invasion. These data suggest that even in the absence of PKR, Salidroside was still able to induce cell death and inhibit cell survival but at a lower level. Based on these data, it becomes clear to us that the anti-proliferative, anti-invasiveness, and apoptotic effect of Salidroside in H29 cells involves multiple mechanisms in which activation of the PKR/eIF-2 pathway is just one mechanism and other signaling pathways are involved. Besides, it suggests that CRC in HT29 cells is a very complicated process and involves the activation of PKR and other signalling pathways. Furthermore, these data suggest that the activation of caspase-8 by Salidroside is completely PKR-dependent.
Interestingly, the deletion of PKR alone partially reduced cell survival and cell proliferation and partially increased LDH release, cell death, and levels of caspases-3. It also and completely prevented the increase in caspase-8 and phosphorylation of eIF-2 as compared to control cells. Another important finding is that deletion of PKR increased cell migration and invasion, even one could expect that the deletion of PKR will increase cell proliferation and inhibit cell apoptosis (by assuming to act as a tumor suppressor through decreasing eIF-2), these data suggest that the basal levels of PKR in HT29 CRC cells may also play a significant role in HT29 maintaining tumor progression. Such partial tumorigenic effects observed in PKR-deficient cells could be explained by the concomitant reduction in the activation of STAT-3 and NF-B (as discussed later) which normally promote cancer progression and inhibit tumor apoptosis. However, our data still validate that sustained activation of PKR by Salidroside stimulates partial cell apoptosis thus suggesting a PKR to act as a tumor suppressor effect after Salidroside treatment mainly through increasing the activation of eIF-2. It should be also mentioned here that the phosphorylation of eIF-2 is under the control of other kinases rather than PKR including (PKR) -like endoplasmic reticulum kinase (PERK); general control non-derepressible 2 kinase (GCN2), and heme-regulated eIF2a kinase (HRI) [4, 5]. However, since Salidroside was unable to phosphorylate eIF2a in PKR deficient cells, these data suggest that PKR is the most targeted kinase on which Salidroside acts to inhibit eIF-2.
Nonetheless, PKR could also induce cell apoptosis in other mechanisms that are completely independent of eIF-2 including activation of p53, JNK, and p38 MAPK [5]. p53 induces cytochrome C release and activates mitochondria-mediated (intrinsic) cell death by upregulation of the Bcl-2-like protein 4 (Bax) [34]. However, JNK can result in cell apoptosis by increasing the production of jBID that enhances the Smac release from the mitochondria to induce activation of caspase-8 [35]. Besides, both JNK and p38 MAPK induced cell apoptosis by activation of Bcl-2 pro-apoptotic members such as Bim [36]. Additional extra mechanisms by which JNK and p53 acts as inducers of caspase-dependent cell death are well-explained somewhere else [37, 38].
In this study, Salidroside also increased JNK activation and increased protein levels of p53 and p38 MAPK. However, Salidroside was unable to stimulate these apoptotic markers in PKR deficient HT29 cells, thus indicating that Saldiorside-induced activation of p38 MAPK, p53, and JNK is PKR-dependent. Given that caspase-8 is mainly induced by JNK, such a PKR-dependent increase in JNK could explain why the upregulation of caspase-8 was also PKR dependent. Although the effect of Salidroside on MAPK activation was poorly investigated in tumor cell lines, the protective effect of Salidroside on breast and lung cancer cells was mediated by inhibition of p38MAPK which contradicts our findings. Such variation could be related to variation in cell lines, treatment, doses used, and experimental conditions.
On the other hand, cancer is described as an inflammatory disorder where inflammation creates a suitable environment needed for tumor growth, metastasis, and stemness [39]. NF-B and STAT-3 are two related inflammatory transcription factors that play active roles in promoting cancer development, progression, invasion, and metastasis of most solid tumors [40]. It was shown that a cross-talk exists between STAT3 and NF-B where inflammatory cytokines (i.e. IL-6) induce sustained activation of NF-B and STAT3, in turn, promotes the activation of NF-B [41, 42, 43]. In this regard, the tumor suppressor effect of NF-B is mediated by the expression of numerous anti-apoptotic genes such as the members of the B-cell lymphoma-2 (Bcl-2) family, the caspase-8 inhibitor, FLIP, and the inhibitor of apoptosis proteins including c-IAP1/2 and XIAP [40]. Similarly, STAT-3 can stimulate cycle progression by stimulating the expression of CyclnD1, c-Myc and Survivin and inhibit cell apoptosis by upregulating numerous anti-apoptotic protein such as Bcl-2, B-cell lymphoma-2-like 1 (Bcl-XL) and myeloid cell leukaemia sequence (Mcl1) [44, 45, 46, 47]. In addition, NF-B induced angiogenesis and stimulates cancer metastasis by increasing the expression of vascular endothelial growth factor (VEGF) and matrix metallopeptidase (MMPs) [40]. Similarly, STAT-3 induces MMPs and other metastatic mediators such as high-mobility group box 1 (HMGB1), Twist, Vimentin, and Snail [43].
Nevertheless, the activities of NF-B and STAT3 were significantly increased in the colon and CRC [48]. It was shown that that PKR can stimulate the activity of both NF-B and STAT-3 in cancer cells [5]. Indeed, the deletion of PKR in HT29 cells of this study inhibited the nuclear activity of both STAT3 and NF-B which support its regulatory effect on these parameters. As mentioned previously, this could explain why PKR deficient cells showed a partial reduction in cell survival and proliferation and partial activation of cell apoptosis. Hence, given its stimulatory effect on PKR, it is reasonable to think that Salidroside may activate these markers to challenge its anti-apoptotic effect. Unexpectedly, we found significantly lower activities of STAT3 and NF-B in Salidroside-treated control cells. Besides, there was a maximum decrease in the activities of STAT-3 and NF-B in PKR deficient cells co-treated with Salidroside. Based on these data, and in addition to stimulation of PKR/eIF-2/P53/JNK/p38 MAPK pathway, it becomes obvious that inhibition of NF-B and STAT-3 are other mechanisms by which Salidroside induces CRC cell death. This could explain why there was a partial reduction in cell survival and proliferation and partial induction of LDH release, apoptosis, and caspaspe3/8 activation in Salidroside-treated PKR deficient-cells as compared to Salidroside-treated control cells. Supporting our data, the inhibitory role of Salidroside on NF-B and STAT3 was shown in normal and cancer cells, including thyroid cancer, osteosarcoma, and CRC [27, 30, 31, 48, 49]. Although not investigated here, a recent study has shown that the inhibitory role of Salidroside on NF-B and STAT3 involves activation of AMPK and SIRT1 [50].
In conclusion, our study is the first to show that the Salidroside tumor suppressor effect in HT29 CRC cells requires concomitant activation of PKR and inhibition of STAT3 and NF-B. These data are very encouraging for future clinical trials.
Footnotes
Acknowledgments
The authors extend their appreciation to the deanship of Scientific Research at King Khalid University, Abha, KSA for supporting this work under grant number (R.G.P.2/80/41). Also, this work was supported by the Taif University Researchers Supporting Project Number (TURSP-2020/99), Taif University, Taif, Saudi Arabia.
References
1.
SiegelR.L.MillerK.D. and JemalA., Cancer statistics, 2019, CA Cancer J Clin69 (2019), 7–34.
2.
LongA.G.LundsmithE.T. and HamiltonK.E., Inflammation and colorectal cancer, Curr Colorectal Cancer Rep13 (2017), 341–351.
3.
LinS.LiY.ZamyatninA.A.WernerJ. and BazhinA.V., Reactive oxygen species and colorectal cancer, J Cell Physiol233 (2018), 5119–5132.
4.
GarciaM.A.GilJ.VentosoI.GuerraS.DomingoE.RivasC. and EstebanM., Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action, Microbiol Mol Biol Rev70 (2006), 1032–1060.
5.
Garcia-OrtegaM.B.LopezG.J.JimenezG.Garcia-GarciaJ.A.CondeV.BoulaizH.CarrilloE.PeranM.MarchalJ.A. and GarciaM.A., Clinical and therapeutic potential of protein kinase PKR in cancer and metabolism, Expert Rev Mol Med19 (2017), e9.
6.
Gal-Ben-AriS.BarreraI.EhrlichM. and RosenblumK., PKR: A kinase to remember, Front Mol Neurosci11 (2018), 480.
7.
HugonJ.PaquetC. and ChangR.C., Could PKR inhibition modulate human neurodegeneration? Expert Rev Neurother9 (2009), 1455–1457.
8.
WatanabeT.ImamuraT. and HiasaY., Roles of protein kinase R in cancer: Potential as a therapeutic target, Cancer Sci109 (2018), 919–925.
9.
MarchalJ.A.LopezG.J.PeranM.CominoA.DelgadoJ.R.Garcia-GarciaJ.A.CondeV.ArandaF.M.RivasC.EstebanM. and GarciaM.A., The impact of PKR activation: From neurodegeneration to cancer, Faseb J28 (2014), 1965–1974.
10.
ShimadaA.ShiotaG.MiyataH.KamahoraT.KawasakiH.ShirakiK.HinoS. and TeradaT., Aberrant expression of double-stranded RNA-dependent protein kinase in hepatocytes of chronic hepatitis and differentiated hepatocellular carcinoma, Cancer Res58 (1998), 4434–4438.
11.
KwonH.C.MoonC.H.KimS.H.ChoiH.J.LeeH.S.RohM.S.HwangT.H.KimJ.S. and KimH.J., Expression of double-stranded RNA-activated protein kinase (PKR) and its prognostic significance in lymph node negative rectal cancer, Jpn J Clin Oncol35 (2005), 545–550.
12.
PataerA.RasoM.G.CorreaA.M.BehrensC.TsutaK.SolisL.FangB.RothJ.A.WistubaI.I. and SwisherS.G., Prognostic significance of RNA-dependent protein kinase on non-small cell lung cancer patients, Clin Cancer Res16 (2010), 5522–5528.
13.
GuoC.ShaoR.CorreaA.M.BehrensC.JohnsonF.M.RasoM.G.PrudkinL.SolisL.M.NunezM.I.FangB.RothJ.A.WistubaI.I.SwisherS.G.LinT. and PataerA., Prognostic significance of combinations of RNA-dependent protein kinase and EphA2 biomarkers for NSCLC, J Thorac Oncol8 (2013), 301–308.
14.
TeradaT.MaetaH.EndoK. and OhtaT., Protein expression of double-stranded RNA-activated protein kinase in thyroid carcinomas: correlations with histologic types, pathologic parameters, and Ki-67 labeling, Hum Pathol31 (2000), 817–821.
15.
BlalockW.L.BavelloniA.PiazziM.TagliaviniF.FaenzaI.MartelliA.M.FolloM.Y. and CoccoL., Multiple forms of PKR present in the nuclei of acute leukemia cells represent an active kinase that is responsive to stress, Leukemia25 (2011), 236–245.
16.
ChengX.BennettR.L.LiuX.ByrneM. and Stratford MayW., PKR negatively regulates leukemia progression in association with PP2A activation, Bcl-2 inhibition and increased apoptosis, Blood Cancer J3 (2013), e144.
17.
ChengX.ByrneM.BrownK.D.KonoplevaM.Y.KornblauS.M.BennettR.L. and MayW.S., PKR inhibits the DNA damage response, and is associated with poor survival in AML and accelerated leukemia in NHD13 mice, Blood126 (2015), 1585–1594.
18.
OshimaM. and IwamaA., Nuclear, not cytoplasmic, PKR maneuvers in AML, Blood126 (2015), 1523–1524.
19.
GilJ.GarciaM.A. and EstebanM., Caspase 9 activation by the dsRNA-dependent protein kinase, PKR: Molecular mechanism and relevance, FEBS Lett529 (2002), 249–255.
20.
C. von Roretz and GallouziI.E., Protein kinase RNA/FADD/ caspase-8 pathway mediates the proapoptotic activity of the RNA-binding protein human antigen R (HuR), J Biol Chem285 (2010), 16806–16813.
21.
WilliamsB.R., Signal integration via PKR, Sci STKE2001 (2001), re2.
22.
GohK.C.de VeerM.J. and WilliamsB.R., The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin, Embo J19 (2000), 4292–4297.
23.
PatersonI. and AndersonE.A., Chemistry. The renaissance of natural products as drug candidates, Science310 (2005), 451–453.
24.
YinD.YaoW.ChenS.HuR. and GaoX., Salidroside, the main active compound of Rhodiola plants, inhibits high glucose-induced mesangial cell proliferation, Planta Med75 (2009), 1191–1195.
25.
ZhaoG.ShiA.FanZ. and DuY., Salidroside inhibits the growth of human breast cancer in vitro and in vivo, Oncol Rep33 (2015), 2553–2560.
26.
LvC.HuangY.LiuZ.X.YuD. and BaiZ.M., Salidroside reduces renal cell carcinoma proliferation by inhibiting JAK2/STAT3 signaling, Cancer Biomark17 (2016), 41–47.
27.
ShangH.WangS.YaoJ.GuoC.DongJ. and LiaoL., Salidroside inhibits migration and invasion of poorly differentiated thyroid cancer cells, Thorac Cancer10 (2019), 1469–1478.
28.
El-KottA.F., M.A. Al-Kahtani and ShatiA.A., Calycosin induces apoptosis in adenocarcinoma HT29 cells by inducing cytotoxic autophagy mediated by SIRT1/AMPK-induced inhibition of Akt/mTOR, Clin Exp Pharmacol Physiol46 (2019), 944–954.
29.
ShatiA.A. and AlfaifiM.Y., Trans-resveratrol inhibits tau phosphorylation in the brains of control and cadmium chloride-treated rats by activating PP2A and PI3K/Akt induced-inhibition of GSK3beta, Neurochem Res44 (2019), 357–373.
30.
SunK.X.XiaH.W. and XiaR.L., Anticancer effect of salidroside on colon cancer through inhibiting JAK2/STAT3 signaling pathway, Int J Clin Exp Pathol8 (2015), 615–621.
31.
HuangL.HuangZ.LinW.WangL.ZhuX.ChenX.YangS. and LvC., Salidroside suppresses the growth and invasion of human osteosarcoma cell lines MG63 and U2OS in vitro by inhibiting the JAK2/STAT3 signaling pathway, Int J Oncol54 (2019), 1969–1980.
32.
WangJ.LiJ.Z.LuA.X.ZhangK.F. and LiB.J., Anticancer effect of salidroside on A549 lung cancer cells through inhibition of oxidative stress and phospho-p38 expression, Oncol Lett7 (2014), 1159–1164.
33.
FanX.J.WangY.WangL. and ZhuM., Salidroside induces apoptosis and autophagy in human colorectal cancer cells through inhibition of PI3K/Akt/mTOR pathway, Oncol Rep36 (2016), 3559–3567.
34.
ChipukJ.E.KuwanaT.Bouchier-HayesL.DroinN.M.NewmeyerD.D.SchulerM. and GreenD.R., Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis, Science303 (2004), 1010–1014.
35.
DengY.RenX.YangL.LinY. and WuX., A JNK-dependent pathway is required for TNFalpha-induced apoptosis, Cell115 (2003), 61–70.
36.
CaiB.ChangS.H.BeckerE.B.BonniA. and XiaZ., p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65, J Biol Chem281 (2006), 25215–25222.
37.
PlotnikovA.ZehoraiE.ProcacciaS. and SegerR., The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation, Biochim Biophys Acta1813 (2011), 1619–1633.
38.
CargnelloM. and RouxP.P., Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases, Microbiol Mol Biol Rev75 (2011), 50–83.
39.
BalkwillF. and MantovaniA., Inflammation and cancer: Back to virchow? Lancet357 (2001), 539–545.
40.
XiaY.ShenS. and VermaI.M., NF-kappaB, an active player in human cancers, Cancer Immunol Res2 (2014), 823–830.
41.
HeG. and KarinM., NF-kappaB and STAT3 - key players in liver inflammation and cancer, Cell Res21 (2011), 159–168.
42.
YuH.PardollD. and JoveR., STATs in cancer inflammation and immunity: a leading role for STAT3, Nat Rev Cancer9 (2009), 798–809.
43.
LeeH.JeongA.J. and YeS.K., Highlighted STAT3 as a potential drug target for cancer therapy, BMB Rep52 (2019), 415–423.
44.
Catlett-FalconeR.LandowskiT.H.OshiroM.M.TurksonJ.LevitzkiA.SavinoR.CilibertoG.MoscinskiL.Fernandez-LunaJ.L.NunezG.DaltonW.S. and JoveR., Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells, Immunity10 (1999), 105–115.
45.
Epling-BurnetteP.K.LiuJ.H.Catlett-FalconeR.TurksonJ.OshiroM.KothapalliR.LiY.WangJ.M.Yang-YenH.F.KarrasJ.JoveR. and LoughranT.P., Jr., Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression, J Clin Invest107 (2001), 351–362.
46.
CorvinusF.M.OrthC.MorigglR.TsarevaS.A.WagnerS.PfitznerE.B.BausD.KaufmannR.HuberL.A.ZatloukalK.BeugH.OhlschlagerP.SchutzA.HalbhuberK.J. and FriedrichK., Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth, Neoplasia7 (2005), 545–555.
47.
LinL.LiuA.PengZ.LinH.J.LiP.K.LiC. and LinJ., STAT3 is necessary for proliferation and survival in colon cancer-initiating cells, Cancer Res71 (2011), 7226–7237.
48.
LiH. and ChenC., Inhibition of autophagy enhances synergistic effects of Salidroside and anti-tumor agents against colorectal cancer, BMC Complement Altern Med17 (2017), 538.
49.
WangJ.PanY.CaoY.ZhouW. and LuJ., Salidroside regulates the expressions of IL-6 and defensins in LPS-activated intestinal epithelial cells through NF-kappaB/MAPK and STAT3 pathways, Iran J Basic Med Sci22 (2019), 31–37.
50.
XuF.XuJ.XiongX. and DengY., Salidroside inhibits MAPK, NF-kappaB, and STAT3 pathways in psoriasis-associated oxidative stress via SIRT1 activation, Redox Rep24 (2019), 70–74.