
Abstract
BACKGROUND:
Circular RNAs (circRNAs) are endogenous RNAs that have a covalent closed cycle configuration. circRNAs have been found to be differentially expressed in many human cancers. Therefore, circRNAs may be ideal biomarkers for the diagnosis and treatment of cancer. However, we know very little about the function of circRNAs in nasopharyngeal carcinoma (NPC). The purpose of this study was to evaluate the circRNA expression profiles in NPC.
METHODS:
We utilized high-throughput RNA sequencing (RNA-Seq) to evaluate the circRNA expression profile in NPC A total of 13,561 unique circRNA candidates were detected. Selection of aberrantly expressed circRNAs was carried out using a
RESULTS:
In NPC tissues, we found that 73 circRNAs were downregulated and 59 were upregulated. The top 12 candidate circRNAs were selected from several vital NPC pathways such as the human papillomavirus and Epstein-Barr virus infection signaling pathways (hsa05165 and hsa05169, respectively), Hepatitis B (hsa05161), and the Ras signaling pathway (hsa04014). A network map of circRNA-miRNA interactions of 12 differentially expressed circRNAs was built. Hsa_circ_0007637 expression distinguished NPC tissues from paired healthy tissues and NPC cell lines (HNE1 6-10B, 5-8F, CNE-2, and so on) from a normal epithelial (NP460) cell line.
CONCLUSIONS:
In this study, we investigated the profiles of differentially expressed circRNAs in NPC, and our results show that hsa_circ_0007637 may be a biomarker for NPC and play a role in its development. This observation-based research identified dysregulated circRNAs in NPC, which may assist in the development of biomarkers for this disease. Further studies on the mechanisms and functions of these circRNAs may promote our understanding of NPC tumorigenesis.
Background
Nasopharyngeal carcinoma (NPC) is one of the most common malignant epithelial tumors and is located in the nasopharynx. It exhibits high invasiveness and metastatic potential, and occurrence rates are specifically high in northern Africa, Southeast Asia, and southern China [1]. In the high-risk areas of South China, the occurrence rate of NPC is 11.3 per 100,000 women and 27.2 per 100,000 men, 50 times higher than that of the United States, Europe, and other countries [2, 3]. When detected early, radiotherapy is an effective treatment, but most patients (75–90%) with NPC are diagnosed with advanced cancer, in which the cancer cells have spread regionally or locally, impeding effective therapy. These patients have a high risk of tumor metastasis and a high rate of disease recurrence [4]. The clinical therapy outcomes of NPC patients is poor, with a 5-year survival rate of only 35% according to one cohort study [5]. Recently, molecular therapy has become a research focus with the advent of precision medicine [6]; hence, a better understanding of biomarkers connected with NPC can play a considerable part in disease perception and prognosis. It can also facilitate the development of stratification, nettargeted treatments and improved prognosis.
In the past few years, although the molecular markers known so far are mostly protein-coding genes, improved clinical outcomes for biomarkers detected in patient subgroups and the development of targeted treatments has been led by validation of biomarkers in specific types of tumors. Some genetic changes related to the pathogenesis of NPC (e.g., LMP1 COX-2 RAS, and EBNA1 mutations) have been assessed as prognostic or diagnostic markers [7, 8]. Nevertheless, less than 2% of the human genome encodes proteins, and the majority of the human transcriptome is composed of noncoding RNAs (ncRNAs). The dysregulation of noncoding RNAs, such as long noncoding RNAs (lncRNAs) and miRNAs, plays a considerable role in many physiological processes in cancers [9, 10]. Recently, a novel class of noncoding RNAs called circRNAs has become an intense focus of study. circRNAs are closed circular RNA molecules which are generated by selective cleavage of premature mRNA. circRNAs are produced by linking the 5’ and 3’ ends generated by backsplicing of linear RNA [11] and are widely found in diverse cell species and types, especially in mammals. Accumulating evidence suggests that circRNAs are specifically expressed at different stages and in different tissues, suggesting that they play a part in many pathophysiological and physiological processes [12, 13, 14]. Therefore, circRNAs may be promising cancer biomarkers [15, 16]. Nevertheless, until now, we know very little about the circRNAs associated with NPC. In this study, we identified circRNA profiles in NPC patients by RNA-Seq and verified dysregulated circRNAs in NPC tissues. Additionally, to accelerate our understanding of the pathogenesis of NPC, we analyzed the correlation between these circRNAs and their microRNA-mRNA interactions. We carried out bioinformatics analyses to predict the possible functions of these circRNAs in NPC and lay the groundwork for prospective NPC-related circRNA studies. Critical insight into the biology of cancer progression and development may be gained by confirmation of the cancer-associated circRNAs identified here.
Methods
Clustering and grouping of NPC samples by pathological classification
Thirty pairs of NPC and adjacent non-tumorous (NT) tissues were collected from patients at the Department of Head Neck Surgery, Otolaryngology, Shenzhen Second People’s Hospital between Jan 2018 and May 2018 (Table S1). The NT tissues were about 5 mm far away from the NPC tissues. The volume of all the NPC and NT tissues are about 2 mm
Representative H&E staining image of nasopharyngeal carcinoma (NPC) and adjacent non-tumorous (NT) tissues.
(A) A volcano map of differentially expressed circRNAs in NPC. The red and green dots show differentially expressed circRNAs that have statistical significance. (B) A volcano map displaying the general characteristics of circRNAs that were differentially expressed between NPC and paired nearby NT tissues. The greater the deviation from the diagonal point, the greater the circRNA expression difference between NPC and paired nearby NT tissues. (C) Stratified cluster analysis of differentially expressed circRNAs. The purple and yellow colors show low and high manifestation, respectively. (D) CircRNAs have diverse genomic allocation because they can originate from intergenic areas, introns, or exons. Accordingly, the pie chart displays the mapping of circRNAs onto the genome. The majority of circRNAs derive from exonic areas, followed by introns and intergenic regions.
RNA isolation from each sample was carried out using RNase/DNase free water in an RNA-dedicated workspace. In short, based on the manufacturer’s instructions, total RNA was isolated from NPC tissues and stored in TRlzol reagent (Invitrogen, Carlsbad, CA, USA) to pair with nearby NT tissues. Total RNA from each sample was washed twice in 75% ethanol, then air-dried and re-suspended in RNase/DNase free water. Following this, the Turbo DNase Kit (Ambion, Foster City, CA, USA) was used to degrade the remaining DNA.
Utilizing a NanoDrop DN2000 spectrophotometer (NanoDrop, Wilmington, DE, USA), we conducted quality assurance and quantified the total RNA, and we accepted OD260/280 ratios between 1.8 and 2.1. In order to enrich for circRNAs, we removed linear RNAs from the total RNA of each specimen after treating with RNaseR (Epicentre, Madison, WI, USA).
Based on the instructions of the manufacturer, RNA Stranded by ribosomal RNA-depleted RNAs and TruSeq Stranded Total RNA Library Prep Kit (Illumina, San Diego, CA USA) was utilized to establish RNA libraries. We denatured the libraries into single-stranded DNA molecules, which were acquired on Illumina flow cells and extended in situ as clusters. Sequencing was performed for 150 cycles on an Illumina HiSeq 4000 sequencer.
High quality trimmed reads(Supplementary methods)were used to identify circRNAs. The reading of each TopHat-fusion reads, which includes TopHat Fusion mapping and TopHat mapping [1], was used to estimate the circRNAs expression level. Hierarchical clustering and volcano maps were used to detect differentially expressed circRNAs at statistical significance between NPC and nearby NT tissues. The statistical data of differences was evaluated by
miRNA prediction and circRNAmiRNA network building
circRNAs include miRNA binding motifs and can be used as miRNA sponges In order to estimate the possible functions of circRNAs in NPC, the MiRanda algorithm in Arraystar miRNA target prediction software was utilized to predict potential circRNA-miRNA interactions for the top 12 upregulated and downregulated circRNAs identified by Illumina sequencing. Based on the predicted miRNA binding motifs, we established a circRNAmiRNA network to probe miRNA response elements (MREs), and Cytoscape was utilized to analyze interactions in the network. The size of node indicates the number of predicted miRNAs that functionally interacted with each circRNA.
The parameters which were utilized for the Arraystar MRE analysis have been formerly published and include the following [37]: (1) nucleotides that are close to pairing with the 3’ pairing sequence of the miRNA; (2) a degree of similarity between AU-richness and seed sequence; and (3) seed forms among nucleotides 2 to 7 by using seed-sequence complementarity. Cytoscape 3.01 was utilized to establish the circRNAmiRNA interaction network map.
Bioinformatics analysis to predict cancer-related circRNA-miRNA target genes
There are many online bioinformatics platforms that can be utilized to forecast the relationship between circRNA-miRNA and target genes. First, we utilized the DIANA-miRPath v.3 platform to confirm miRNA candidates in Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) cancer-relevant pathways. The IntaRNA platform, which can predict interactions between RNA molecules, was utilized to match candidate circRNAs with the seed sequences of forecasted target miRNA candidates. Second, after circRNA target miRNA interactions were confirmed, we utilized the DIANA-TarBase 7.0 database to authenticate the top 12 cancer-relevant target genes of each target miRNA candidate.
Details of primer pairs used in the analysis of circRNA expression by qRT-PCR
Details of primer pairs used in the analysis of circRNA expression by qRT-PCR
CircID
Base on the bioinformatics analysis of cancer-related circRNA-miRNA target genes, we ranked and identified the 12 circRNAs closely related to cancer miRNA, and verified the top five circRNAs (hsa_circ_0007637, hsa_ circ_0001516, hsa_circ_0000713, hsa_circ_0008085 and hsa_circ_0036353) by qRT-PCR. Based on the instructions of the manufacturer and RNase R treatment, cDNA was synthesized by reverse transcribing qualified total RNA from five chosen circRNAs utilizing the SuperScript first chain synthesis system for RT-PCR kit (Invitrogen). We utilized SYBR-Green Premix Ex Taq (Takara Bio, Nojihigashi, Kusatsu, Japan) to carry out qRT-PCR on the ABI PRISM 7500 Sequence Detection System (Applied Biosystems, Life Technologies, Waltham, MA, USA). With the reagents below, the reaction was carried out in a 20
Results
Properties of the human NPC samples
High throughput sequencing was utilized to test six samples of NPC tissues and comparative nearby NT tissues(A1-A3,B1-B3), which were identified based on histopathological examination. Detailed information on patient characteristics is provided in Fig. 1. Besides, another 27 pairs of samples were used for genetic identification. A detailed list can be found in Table S1.
CircRNA expression profiles in NPC and NT tissues
In order to analyze circRNA expression in NT and NPC tissues, we carried out high throughput sequencing. We identified 13,561 circRNAs in total, and approximately 82% of reads mapped to exons (Fig. 1d). Student’s
(A) GO terms significantly enriches differentially expressed genes. The bar graph displays the enrichment fraction of the first 10 statistically significantly enriched GO terms (-log10 [
(A) A network of circRNA-miRNA interactions in NPC. The network diagram contains 12 significantly altered circRNAs, which are shown as blue dots, that identified in the circRNA-miRNA network predictive analysis. The purple dots around the blue are predicted miRNAs that interact with the associated circRNA. (B) Predicted miRNA-circRNA interactions. The cancer-relevant miRNA is noted by green dots, and the number of cancer-relevant miRNAs per circRNA is shown as a blue dot and was calculated in Additional file 1: Table S3.
(A) Hsa_circ_0007637 expression levels of 30 paired NPC tissues and comparative nearby NT tissues. We used the 2(-Ct) method to verify circRNA expression by qRT-PCR. The data are shown as the mean 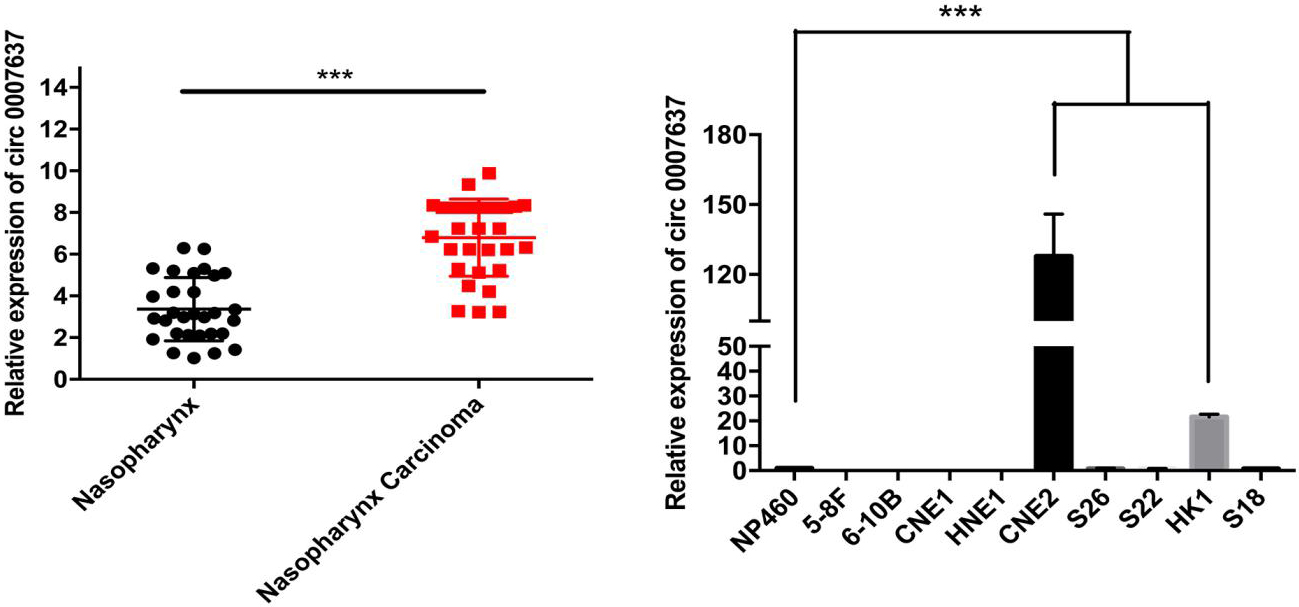
In order to identify NPCrelevant circRNAs, we carried out KEGG pathway analyses of the genes that produced differentially expressed circRNAs. In the ‘biological process’ term, the most enriched GO terms were binding molecular function classifications and cellular component (
Based on this, we ranked and identified the top 12 candidate cancer-related circRNAs from the GO and KEGG analyses (Table S3) to determine whether these circRNAs were expressed in NPC.
Accumulating evidence indicates that circRNAs contain corresponding miRNA binding sites and have an influence on miRNA-mediated gene expression regulation [16, 17]. Competitive endogenous RNAs (ceRNAs) contain shared MREs, such as circRNAs, long noncoding RNAs and mRNAs, and could compete for miRNA binding [18]. Therefore, we investigated miRNAs potentially associated with the top 12 circRNAs in KEGG utilizing Arraystar circRNA target prediction software. Based on the predicted MREs, we established a circRNA-miRNA interaction network plot (Fig. 4A). A bioinformatics platform was utilized to contrast the associations among cancer-relevant miRNAs and miRNAs that interacted with the 12 candidate circRNAs to better understand the molecular mechanisms underlying the differential expression of circRNAs in NPC. Utilizing Cytoscape, we established a cancer-relevant circRNA-miRNA target gene map (Fig. 4B). Additional file 1: Table S3 displays the number of each dysregulated circRNA that interact with the cancer-related miRNAs noted in Fig. 5. This analysis revealed circRNAs involved in highly expressed pathways related to NPC and nearly half of the circRNAmiRNA interactors were cancerrelated factors (176/368) (Additional file 1: Table S3). We confirmed several cancer-associated pathways that may be feasible targets for these circRNA-miRNA-mRNA interactions in NPC (Fig. 4B).
Validation of circRNAs by qRT-PCR
Based on the circRNA expression profiles, and bioinformatics analysis of cancer-related circRNA-miRNA target genes, we selected the top five circRNAs for further validation. In previous experiments, we found that only Has_circ_0007637 showed significant differences between NPC tissues and nearby NT tissues the results of the other four candidate circRNAs were also shown in Fig. S3. We first observed that the expression level of Has_circ_0007637 was upregulated in 30 NPC tissue samples compared to nearby NT tissues by qRT-PCR (
Discussion
CircRNAs are a type of non-coding RNA that have been neglected as transcriptional noise in eukaryotes for the last 30 years [19, 20]. Current studies have clearly shown that circRNAs are diverse, stable, and conserved RNA molecules [21]. Compared to IncRNAs, miRNAs and mRNAs, circRNAs are potential biomarkers for the diagnosis and prognosis of diseases due to their stability [22]. Convincing evidence suggests that circRNAs can regulate gene expression through many mechanisms. For example, circRNAs serve as miRNA sponges and competitively bind miRNAs to regulate gene expression [18, 23]. The transcription of exon-intron circRNAs occurs via RNA-RNA interactions in the nucleus [24, 25]. Some reports have shown that circRNAs could be translated into proteins [26, 27]. Studies show that circRNAs play a considerable role in transcriptional and posttranscriptional gene expression and can serve as ideal markers for disease diagnosis. Many circRNAs have been shown to be dysfunctional in cancer [28, 29]. For example, Wan et al. [30] showed that circ-ITCH is overexpressed in lung cancer tissues and serves as a miR-7 and miR-214 sponge to restrain the Wnt/
In this study, in order to accelerate our understanding of the pathogenesis of NPC, sequencing was used to assess the expression of circRNAs in NPC. We identified a total of 13,562 circRNAs, of which about 82% mapped to genomic exons. We also probed the possible function of circRNAs, which may act as biomarkers for the diagnosis and prediction of disease. Statistically significant differentially expressed circRNAs were identified (Q-value
Previous molecular studies of NPC have primarily concentrated on genomic analyses. In last few years, studies have also concentrated on noncoding RNAs in NPC. Studies on circRNAs in NPC are limited, and research based on circRNA-miRNAmRNA interactions is a rising research field [36].
To our knowledge, few studies have been conducted to evaluate circRNAs in NPC. Our study comprehensively profiled the possible interactions between 12 candidate circRNAs and miRNAs. This search was based on the hypothesis that highly expressed circRNAs may interact with diverse cancerrelevant miRNAs and thus regulate cancerrelevant pathways. We ranked and identified miRNAs for Cancer related circRNAs, and investigated the functions of their target genes using KEGG. We actually performed RT-PCR validation on the Top5 circRNAs. Ultimately, we found that Hsa_circ_0007637 was up-regulated in both of CNE2 and HK1 cell lines and in NPC tissues. It is worth noting that Hsa_circ_0007637 originates from the aberrant mRNA splicing of another well-known cancer-related gene, CREBBP, which has been reported in many types of human cancers including NPC. Therefore, the tumor-specific expression of this circRNA is reasonable.
In summary, our study identified circRNA expression profiles in NPC. Compared to nearby NT tissues, several circRNAs were discovered to be mis-regulated in NPC tissues. qRT-PCR analysis confirmed the RNA-Seq results. Bioinformatics analysis predicted possible roles for these circRNAs and numerous feasible circRNA-miRNA-mRNA-ceRNA networks. This study also suggests potential biomarkers for NPC. Additional studies on the mechanisms and functions of these circRNAs will accelerate our understanding of NPC tumorigenesis. This study also has the following limitations: 1. The sample size is insufficient, and the follow-up research needs to increase the sample size. 2. We have discovered a large number of circRNAs, and their related biological functions and molecular regulatory mechanisms are still unclear, so further in-depth research is needed in the future.
Conclusions
In summary, high-throughput transcriptome sequencing and bioinformatics analysis of NPC may offer feasible therapeutic targets and diagnostic markers for future studies of this disease.
Footnotes
Acknowledgments
This work was supported by National Natural Science Foundation of China (81970875), Shenzhen Science and Technology Innovation Committee (JCYJ2018 0508152528735, JCYJ20180228163446770), Sanming Project of Medicine in Shenzhen (SZSM201612031). National Natural Science Foundation of China (81773257). Shenzhen Municipal Government of China (JCYJ20180507184642475).
Conflict of interest
There is no conflict of interest in this manuscript.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-201731.