
Abstract
BACKGROUND:
Pancreatic cancer is a malignant tumor and its incidence has increased in recent years. Carboxypeptidase E (CPE) is a prohormone/proneuropeptide processing enzyme that has been shown to be associated with tumor growth and invasion in various cancers including pancreatic cancer.
OBJECTIVE:
To understand the molecular mechanism underlying the proliferative effects of CPE in cancer cells.
METHODS:
We down-regulated CPE gene expression in PANC-1 cell, a pancreatic cell line, and investigated mRNA, miRNA, circRNA and lncRNA expression profiling in PANC-1 cells from control group and CPE knock-down group by microarray analysis. We further validated the top 14 differentially expressed circRNAs by qRT-PCR.
RESULTS:
Our results showed that CPE down-regulation caused decreased cell proliferation. The microarray data showed 107, 15, 299 and 360 differentially expressed mRNAs, miRNAs, circRNAs, and lncRNAs, respectively between control group and CPE knock-down group. Of Which, 41 mRNAs, 12 miRNAs, 133 circRNAs, and 262 lncRNAs were down-regulated; 66 mRNAs, 3 miRNAs, 166 circRNAs, and 98 lncRNAs were up-regulated. Bioinformatics analysis showed that the top significantly enriched pathways for the differentially expressed RNAs were related to cancer onset and/or progression, these included p53 signaling pathway, ECM-receptor interaction, focal adhesion and Wnt signaling pathway. We further performed network analysis to assess the mRNA, miRNA, circRNA and lncRNA correlations, and showed that HUWE1, hsa-miR-6780b-5p, has_circ_0058208 and lnc-G3BP1-3:8 were in the core position of the network.
CONCLUSIONS:
Taken together, these results identified potential CPE regulated core genes and pathways for cell proliferation in pancreatic cancer cell, and therefore provide potential targets for the treatment of pancreatic cancer.
Introduction
Carboxypeptidase E (CPE) is a prohormone/ proneuropeptide processing enzyme and was initially identified in endocrine cells [1]. CPE can specifically bind and catalyze the transformation of multiple hormone enzymes into hormones, thus playing an active role in biological processes [2]. CPE exists in 2 forms, soluble CPE and membrane-binding CPE. It plays an important role in regulating endocrine and nervous system balance, and also has certain ability to promote cancer [3, 4]. In recent years, it has been found that CPE is highly expressed in many malignant tumor tissues [5], participates in the process of tumor occurrence and progression [4, 6], and plays an important role in tumor infiltration and metastasis [7].
Pancreatic cancer is the one of the leading cause cancer-related death worldwide and is a malignant tumor with hidden, rapid development and poor prognosis in clinical manifestations. It has high morbidity and mortality in clinic and various etiologies involving genetic factors such as Kras and BRCA1/BRCA2 gene mutations, and risk factors including smoking, obesity, diabetes and chronic pancreatitis [8, 9]. Although the diagnosis and treatment of pancreatic cancer has been improving, pancreatic cancer is still difficult to diagnose in the early stage and the patient’s 5-year survival rate after surgical treatment is low [9]. Therefore, there is an urgent need to better understand the molecular mechanism underlying the pathogenesis of pancreatic cancer and subsequently develops new treatment of pancreatic cancer.
In recent years, a growing number of studies have shown that non-coding RNAs were important in the pathogenesis of many types of cancers including pancreatic cancer. Long chain non-encoded RNA (lncRNA) is a class of non-encoded RNA with a length exceeding 200 nt. LncRNA can regulate gene expression at multiple levels of life activity, thereby affecting biological processes [10, 11]. circRNA is a unique endogenous non-encoded RNA, which played a key role in the regulation of tumor occurrence and development [12, 13]. Micro RNA is a kind of small molecule RNA which is closely correlate with tumorigenesis [14, 15], belongs to non-encoded single-stranded small molecule RNA, and is mainly involved in the regulation of gene transcription level [16]. Although lncRNAs, circRNAs and miRNAs were suggested to be involved in pancreatic cancer pathogenesis and are promising targets for treatment of the disease [17, 18, 19], bioinformatics analysis of the interactive pathways that associated with dysregulated mRNAs and non-coding RNAs is limited.
To better understand the impact of CPE associated transcriptional and epigenetic regulation in pancreatic cancer, we knock-down CPE in PANC-1 cell, a pancreatic cancer cell line, and used microarray to identify differentially expressed mRNAs, miRNAs, lncRNAs and circRNAs between control cells and CPE knock-down cells. GO and KEGG enrichment pathway analyses were performed to investigate the CPE associated pathways for cancer cell proliferation. Finally, CeRNA analysis was used to evaluate lncRNA/circRNA-miRNA-mRNA competitive regulatory networks.
Materials and methods
Cell lines
PC cell lines, panc-1, provided by Peking Union Medical College Hospital. Cells were maintained in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS), streptomycin (100
Transfection of siRNA
To knock down CPE, CPE-siRNA, along with its nonspecific scramble siRNA, NC-siRNA, were purchased from Guangzhou RiboBio (Guangzhou, Guangdong, China). We transfected the siRNA into cells by Lipofectamine 3000 transfection reagent kit according to manufacturer’s instruction (Invitrogen, Carlsbad, CA, USA).
RNA extraction and quantitative RT-PCR
Total RNA was extracted from cultured cells by a TRIzol reagent following the manufacturer’s instructions (Invitrogen). cDNA was obtained reverse transcriptase kit (Beijing Zoman Biotechnology, Beijing, China). The cDNA was amplified and quantified using a lightcycle 96 detection system with the application of SYBR Green I dye. The primers of CPE were TGTCT GACCCCAATCG (forward) and ACTCCTCGGTGT ATCT (reverse); The primers of CPE were ATGGGGAA GGTGAAGGTCGGAG (forward) and TCGCCCCA CTTGATTTTGGAGG (reverse). Expression data were collected from triplicates, subsequently normalized to GAPDH, and calculated as 2
Cell proliferation assay
Cell viability of the PC cell lines was measured using the WST-1 Cell Proliferation Reagent. 4
Identification of differentially expressed genes
The microarray experiments were performed by following the protocol of Agilent technologies Inc at Shanghai Biotechnology Corporation. RNA samples of each group were used to generate fluorescence labeled cRNA (complementary RNA) targets for the SBC human ceRNA array V1.0 (4
Gene ontology analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment pathways
We used the TargetScan and circBase databases to predict the target genes of miRNA and circRNAs. Regarding the target genes of lncRNA, firstly, the genes within the range of 10 kb upstream and downstream of lncRNA were selected as the cis target genes of this lncRNA. Secondly, the gene sequence of the corresponding species in the database is selected to select the sequence with complementarity or similarity by Blast, and the complementary energy between the two sequences is calculated using RNAplex, and the gene that e
Kyoto Encyclopedia of Genes and Genomes (KEGG) were used to perform the enrichment analysis including cellular component (CC), molecular function (MF), biological process (BP), and signaling pathway. The GO-BP terms, GO-CC terms, and GO-MF terms were filtrated by the standard of
ceRNA analysis
ceRNA analysis is a new model of RNA expression regulation, which can explain the regulation network of lncRNA/circRNA-miRNA-mRNA. In this study, targetscan was used to predict the lncRNA and circRNA that could competitively combine with miRNA. Based on the expression value of gene, the regulation network of sponge adsorption of microRNA was established by regression model analysis and seed sequence matching method. The different types of ceRNA mechanisms in the experimental group and the control group were studied to find specific ceRNA. The relationship between circRNA, lncRNA, mRNA, miRNA is established by using the expression value S, which is used to infer whether miRNA regulates circRNA or lncRNA and mRNA, and to find out the relationship between the three.
Statistical analysis
Data were presented as mean
Results
Down-regulation of CPE inhibited PANC-1 cell growth
To investigate the effect of CPE on PANC-1 cell proliferation, PANC-1 cells were transfected with CPE-siRNA (50 nM) to knock down endogenous CPE in the cells. The transfection of scramble-siRNA was set as negative control group. The qRT-PCR showed that endogenous CPE was significantly down-regulated by CPE-siRNA transfection (Fig. 1A). The role of CPE in cell proliferation was investigated by WST-1 assay. The results demonstrated that knocking down CPE significantly inhibited PANC-1 cell proliferation (Fig. 1B).
CPE downregulation decreased cell proliferation in PANC-1 cells. PANC-1 cells were transfected with CPE-siRNA and scramble-siRNA. A. The transcriptional expression of CPE after siRNA transfection measured by RT-PCR. B. Cell proliferation in PANC-1 cells was measured after 24 h, 48 h and 72 h transfection. 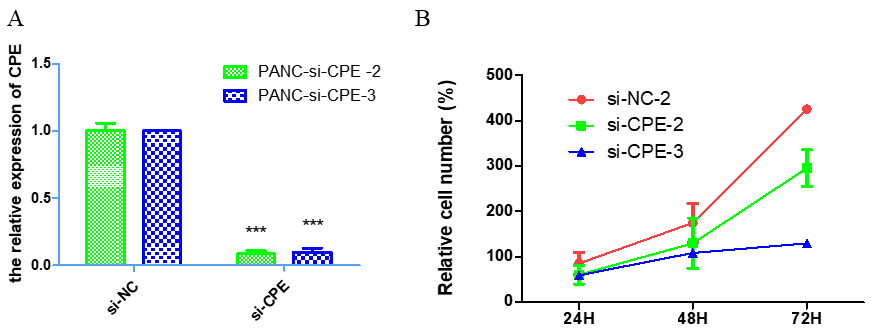
We then used microarray to analyze the DEGs between cells transfected with CPE-siRNA and NC-siRNA. Bioinformatics analysis showed 107 mRNAs, 15 miRNAs, 299 circRNAs and 360 lncRNAs were differentially expressed. Further analysis showed that 41mRNAs, 12miRNAs, 133circRNAs, and 262 lncRNAs were down-regulated, whereas 66 mRNA, 3 miRNA, 166 circRNA, and 98 lncRNA were up-regulated.
To validate the microarray data, we used qRT-PCR to analyze several top differentially expressed circRNAs (Fig. 2A). The results demonstrated that CPE down-regulation significantly increased hsa_circ_0019223, hsa_circ_0087633, hsa_circ_0021692, hsa_circ_ 0009125, hsa_circ_0013502, and hsa_circ_0024109 levels (Fig. 2B–H). In addition, CPE down-regulation significantly decreased hsa_circ_0019721, hsa_circ_ 0091825, hsa_circ_0074239, hsa_circ_0026920, hsa_ circ_0030860, hsa-circ_0091824, hsa_circ_0002449, and hsa_circ_0060318 levels in the PANC-1 cells (Fig. 2I–O). These results were consistent with the microarray data.
circRNA molecule expression pattern under CPE knock-down condition with qRT-PCR validation. (
To know the functional characteristics of the DE mRNAs, miRNAs, circRNAs and lncRNAs, we conducted GO enrichment analyses. As shown in Fig. 3A, the top significant enriched pathways for DE mRNAs included response to alkaloid (GO: 0043279,
Gene ontology functional enrichment analysis for DEGs. A. The top significant GO enrichment pathways for the DE mRNAs genes. B. The top significant GO enrichment pathways for the target genes of DE miRNAs. C. The top significant GO enrichment pathways for the target genes of DE cirRNAs. D. The top significant GO enrichment pathways for the cis target genes of DE lncRNAs. E. The top significant GO enrichment pathways for the trans target genes of DE lncRNAs.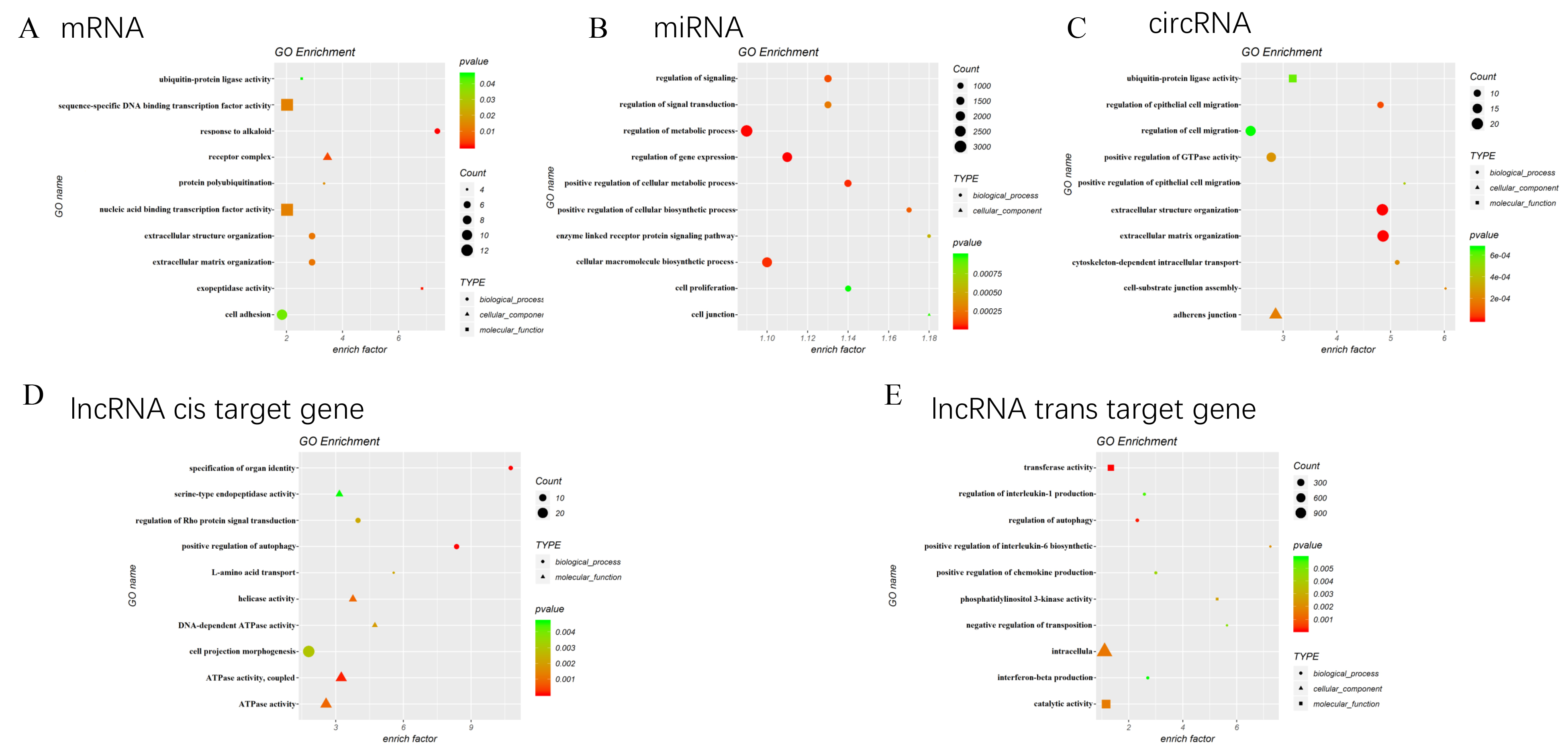
In addition, as shown in Fig. 3B, the top significant enriched pathways for the target genes of DE miRNAs included cell junction (GO: 0030054,
Moreover, as shown in Fig. 3C, the top significant enriched pathways for the target genes of DE circRNAs were extracellular matrix organization (GO: 0030198,
Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis for DEGs. A.The top significant enriched KEGG pathways for DE mRNAs. B. The top significant enriched KEGG pathways for the target genes of DE miRNAs. C. The top significant enriched KEGG pathways for the target genes of DE circRNAs. D. The top significant enriched KEGG pathways for the cis target genes of DE lncRNAs. E. The top significant enriched KEGG pathways for the trans target genes of DE lncRNAs.
Interestingly, the top significant enriched pathways for the cis and trans regulation targets of lncRNAs both included regulation of autophagy (Fig. 3D and E).
We also used KEGG Pathway Enrichment Analysis to understand the functional involvement of DE mRNAs, miRNAs, circRNAs and lncRNAs in CPE regulated cancer cell proliferation. According to the KEGG pathway analysis, there were 4, 3, 2, and 2 mRNAs enriched in pathways in Phagosome, Cell adhesion molecules (CAMs), Pentose and glucuronate interconversions and ECM-receptor interaction, respectively (Fig. 4A).
For the DE miRNA target genes, there were 215, 148, 139, 114, and 86 genes enriched in pathways relate to cancer, Endocytosis, MAPK signaling pathway, Focal adhesion and Wnt signaling pathway, respectively (Fig. 4B).
For the KEGG enrichment analysis of DE circRNA target genes, it showed 8, 7, 6, and 4 genes enriched in Focal adhesion, ECM-receptor interaction, Ubiquitin mediated proteolysis, and Small cell lung cancer, respectively (Fig. 4C).
According to the KEGG pathway-related database, for the lncRNA cis targets, there were 4, 2, 2 and 2 genes enriched in Fc gamma R-mediated phagocytosis, Propanoate metabolism, ABC transporters, and basal transcription factors, respectively (Fig. 4D). Moreover, for the trans targets, there were 23, 20, 18, and 18 genes enriched in Lysosome, NOD-like receptor signaling mTOR signaling pathway, and Jak-STAT signaling pathway, respectively (Fig. 4E).
ceRNA network analysis of the DEGs. Each point represents a gene, line represents the regulated microRNA between genes. A. Represents down. B. Represents up, respectively represent circRNA, lncRNA, and mRNA.
To further understand the functional involvement of DEGs in CPE regulated cancer cell proliferation, the ceRNA analysis was performed to analyze the interactive network of DEGs. As shown in Fig. 5, the size of the circle represents the ability of genes to interact with other genes. For the analysis of mRNAs, HUWE1 (HECT, UBA and WWE domain containing 1, E3 ubiquitin protein ligase) was in the core position, it was down-regulated in siRNA-CPE cell and was predicted to interact with 73 DEG genes to influence the activity of cell. As for the analysis of miRNA, hsa-miR-6780b-5p was in core position and it was predicted to interact with 149 DEG genes. Moreover, the ceRNA analysis revealed that hsa_circ_0058208 was the core circRNA in the network. It was down-regulated and was predicted to interact with 30 DEG genes. As for the analysis of lncRNAs, lnc-NR_023390 and lnc-G3BP1-3:8 were in the core position. Among them, lnc-NR_023390 was up-regulated and it was predicted to interact with 11 DEG genes; lnc-G3BP1-3:8 was down-regulated and it was predicted to interact with 10 DEG genes. According to cis prediction, the target gene of lnc-NR_023390 is ERCC excision repair 6 like 2 [Source: HGNC Symbol-3BAcc: HGNC: 26922], the gene is located on the ninth chromosome and is associated with biological processes such as DNA damage repair. The target gene of lnc-G3BP1-3:8 GM2 ganglioside activator, the gene is located on the fifth chromosome, is a protein-coded gene that it encoded GM2 ganglion glycosides protein, and can Synergy with beta-hexosaminidase A in lysosomes to ensure the normal function of Beta-hexosaminidase A.
Discussion
Pancreatic cancer is a common malignant tumor in clinic, its location is hidden, and early symptoms are not obvious, 80% of patients were at late stages, only a few patients have the opportunity to have surgical resection [9]. CPE is a member of the M14 family metal peptide enzyme, involved in the biosynthesis of peptide hormones and neurotransmitters, but also has a certain role in promoting cancer. Some studies have revealed that the abnormal expression of CPE is closely related to the occurrence and metastasis of tumors [6]. Other studies suggest that the expression of CPE in rectal cancer and breast cancer tissues and cell lines increased significantly, and the high expression of CPE can increase cell proliferation [7, 20]. The results of the present study showed that knockdown of CPE slows cell proliferation in pancreatic cancer cells. Therefore, CPE may be involved in the occurrence and development of pancreatic cancer. We further used microarray to explore potential regulatory mechanism underlying the effect of CPE. Bioinformatics analysis identified CPE associated DE mRNAs, miRNAs, lncRNAs and circRNAs in the PANC-1 cells. In order to gain insight into the functional characteristics of identified DEGs, we conducted GO and KEGG pathway enrichment analysis, ceRNA network analysis to analyze the key genes and pathways for the effect of CPE on pancreatic cancer cell proliferation. ceRNA analysis is a new model of RNA expression regulation, ceRNA means competing endogenous RNA, lncRNA and circRNA can competitively combine with miRNA, which in turn inhibits the expression of miRNA and its negative regulation of downstream target genes.
In the ceRNA network analysis, the core gene were HUWE1 mRNA, lncNR-023390, lnc-G3BP1-3:8, has-circ-0058208, and has-miR-6780b-5p. Ubiquitin-linked enzyme is one of the key enzymes in ubiquitin proteasome system, which is mainly responsible for target protein identification and the regulation of the activity of ubiquitin system. HUWE1 is a class of ubiquitin-linked enzymes with HETC (homologous to E6AP C terminus) functional domains [21, 22]. Previous studies found that HUWE1 was involved in apoptosis, genomic DNA damage, Tumor suppressors regulation, nerve cell differentiation and nerve cell proliferation and other important physiological processes [23, 24, 25]. HUWE1 has also been suggested to be involved in regulating cancer-related molecular activity and pathways, including regulation of MYC transcription activity, P53 activity, and P53 independent anti-proliferation pathways [26, 27, 28].
miRNAs are involved in many important physiological and pathological conditions, and its biological functions include participation in embryonic development, cell proliferation, apoptosis and differentiation [16]. Some studies have confirmed abnormal expression of specific miRNAs may lead to the occurrence of various human diseases, including the occurrence and development of tumors [14, 15, 29]. In our present study the core miRNA is hsa-miR-6780b-5p, the miRNA can be used as a biomarker for adult imported falciparum malaria [30]. Interestingly, bioinformatics analysis indicates that hsa-miR-6780b-5p has a potential to regulate the expression of 1237 genes. However, ceRNA analysis showed that hsa-miR-6780b-5p had potential interaction with 149 DEG genes in PANC-1 cells, suggesting a competition of hsa-miR-6780b-5p with other RNAs for the regulation of gene expression in the cells.
LncRNA have a variety of molecular functions, including regulating transcription, regulating protein activity, playing a structural or organizational role, changing RNA processes, and multi-dimensional expression regulation as a precursor to miRNA, participating in chromosome silencing and modification, transcription activation and interference, activating Proto-oncogene, promoting tumor proliferation, metastasis and many other biological functions [10, 11]. In our present study the core lncRNAs are lncNR-023390 and lnc-G3BP1-3:8. According to cis projection, the target gene for lnc-NR_023390 is ERCC6L2, with its full name ERCC excision repair 6 like 2 [Source: HGNC Symbol-3BAcc: HGNC: 26922], which is located on the ninth chromosome, related to biological processes such as the repair of DNA damage [31]. According to cis prediction, lnc-G3BP1-3:8 ’s target gene is GM2A, the full name is GM2 ganglioside activator, the gene is a protein-coded gene, which is located in the fifth chromosome, encodes GM2 ganglion glycosides protein [32], and Synergies with Beta-hexosaminidase A in lysosomes to ensure the normal function of Beta-hexosaminidase A [33]. Another study showed that GM2A played a role in cancer cell migration [34].
circRNA is a covalent closed non-encoded RNA that can regulate gene expression in eukaryotic organisms. Many circRNAs have organization and timing specificity, and are closely related to physiological conditions and tumor occurrence [35, 36]. circRNA is not easily degraded by nucleic acid excision enzyme, can exist stably in vivo, has a high degree of conservatism, indicating that circRNA has the obvious advantage of becoming a new diagnostic marker. Recent studies have shown that circRNA can play an important role in a variety of cancers, playing the role of a cancerous or anticancer gene in the development of cancer, thus acting as a biomarker for tumors [12, 13, 37]. In our present study, the core circRNA is has-circ-0058208, which comes from the mother Gene TNS1. TNS1, is a protein that binds to actin in the adhesion plaque of the binding site of cells and extracellular matrices. With SH2 functional domain, TNS1 can phosphorylate tyrosine, maintain the tension of the microfilament anchor point, and also can participate in signal transduction and link the signal transmission system to the cytoskeleton. Furthermore, previous studies have shown the elevated expression of TNS1 in cancer
The functional involvement of CPE-related transcriptional and epigenetic changes in PANC-1 cells may involve the Wnt signaling pathway, since it has been reported that CPE regulated cell proliferation and migration via Wnt signal pathway [38, 39], and studies showed that HUWE1 influenced cellular functions through Wnt signaling pathway [40, 41]. Our analysis further showed that lnc-G3BP1-3:8 could competitively interact with hsa-miR-5194, thus influencing the expression of ELL2, which has been demonstrated to suppress canonical Wnt signaling in the cytoplasm [42]. Additionally, ceRNA analysis suggested that hsa_circ_0058208 could competitively interact with hsa-miR-5581-5p, leading to the transcriptional regulation of LHX1, which was shown to modulate the activity of the Wnt signaling pathway [43]. Furthermore, according to ceRNA analysis, hsa-miR-6780b-5p has the potential to interact with HOXA10, which influenced Wnt signaling pathway by regulating
Although our results revealed CPE associated transcriptional and epigenetic dysregulations in cancer cells, there are several limitations in this study. The first limitation is that the results from this study were based on a cell line, but immortalized cells may present mutations and functions differently from in vivo due to multiple immortalization processes. Secondly, it is known that methylation processes invariably occur during cancer onset and/or progression, and therefore methylation analysis and proteomic analysis would probably useful to understand molecular mechanisms underlying the proliferative effects of CPE in cancer cells. In addition, the combination of RNA analysis with DNA analysis, such as mate pair sequencing and ATAC-seq may provide more insights into the role of CPE in cancer.
In conclusion, we explored the CPE associated DE mRNAs, miRNAs, lncRNAs and circRNAs, and identified potential CPE regulated core genes and pathways for cell proliferation in pancreatic cancer cell, and therefore provide potential targets for the treatment of pancreatic cancer.
Footnotes
Acknowledgments
This work was supported by the CAMS Innovation Fund for Medical Sciences (2016-I2M-3-005), the National Natural Science Foundation of China (81703492), and the Beijing Natural Science Foundation (7182092).