
Abstract
BACKGROUND:
Uveal melanoma (UM) is the most common primary intraocular tumor in adults, which has a high mortality rate and worse prognosis. Therefore, early potential molecular detection and prognostic evaluation seem more important for early diagnosis and treatment.
METHODS:
Gene expression data were obtained from The Cancer Genome Atlas-Uveal melanomas database. Survival genes were identified by univariate analysis and were regarded to be associated with the overall survival of UM patients. Then, pathway enrichment analysis of these survival genes was performed. Robust likelihood-based survival model and multivariate survival analysis were conducted to identify more reliable genes and the prognostic signature for UM survival prediction. Two internal datasets and another two UM datasets from Gene Expression Omnibus (GEO) were used for the validation of prognostic signature.
RESULTS:
Firstly, 2,010 survival genes were screened by univariate survival analysis. GO and KEGG analysis revealed that these genes were mainly involved in pathways such as mRNA processing, RNA splicing, spliceosome and ubiquitin mediated proteolysis. Secondly, a six-gene signature was identified by Robust likelihood-based survival model approach. The gene expression of the six genes can successfully divide UM samples into high- and low-risk groups and have strong survival prediction ability. What’s more, the expression of six genes was compared in 80 healthy adipose tissue samples obtained from GTEx (Genotype-Tissue Expression) database and further validated in internal datasets and GEO datasets, which also can predict UM patient survival.
CONCLUSIONS:
The six genes (SH2D3A, TMEM201, LZTS1, CREG1, NIPA1 and HIST1H4E) model might play a vital role in prognosis of UM, which should be helpful for further insight into the treatment of uveal melanoma.
Background
Uveal melanoma (UM) is the most common malignant tumor in adults, which originates from uveal melanocytes and has a high rate of metastasis. The incidence of 6–7 cases per million and nearly 50% of patients with UM develop metastases after diagnosis [1, 2, 3, 4]. The most common metastatic site is the liver, followed by the lung and soft tissues. Although treatments such as chemotherapy, radiotherapy and excision have been used to cure primary tumors and prevent local recurrence, the 5-year survival rate still very low and there is no effective treatment for metastatic UM [5, 6]. Furthermore, although patients with UM are diagnosed in early stage, the suitable treatments can’t implemented in time. The prognosis of many patients remain uncertain and the best treatment after relief is unclear. Therefore, is important to clarify the survival events of UM and identify new prognostic factors and therapy targets.
Cancer, as we known, is a genetic disease characterized by genomic instability and the progressive accumulation of genetic abnormalities or mutations. Nowadays, gene expression microarray of tumor has been widely used in cancer molecular mechanisms and prognosis prediction [7]. Microarray analysis is considered as a promising tool for generating gene expression data on a genomic scale [8], which can be utilized to identify molecular subtypes, discover progression markers and construct models with different prognosis [9]. However, survival research on expression profile of UM was rare, which limited the understanding of critical genes associated with the occurrence and prognosis of these diseases. Such researches could help to solve further molecular mechanisms involved in UM and provide insight into new methods of prevention and treatment.
Recently, bioinformatics has been extensively used in the field of tumor biology. Some public databases, such as TCGA (The Cancer Genome Atlas), GEO (Gene Expression Omnibus) databases are open to researchers [10]. Therefore, in this study, the original data of 80 UM samples downloaded from the Cancer Genome Atlas-Uveal melanomas (TCGA-UVM) were analyzed. By implementing univariate survival analysis and robust likelihood-based survival modelling approach [11, 12]. Six genes regarded as potential prognosis markers were identified. Moreover, the prognostic marker was validated in two independent internal datasets and external datasets (GSE42656 and GSE84976) from GEO. With this model, patients with at least six of these genes differentially expressed can be predicted as high risk of mortality with UM. Our research might provide important potential genes as biomarkers for the prognosis and treatment [13, 14].
Materials and methods
RNA and clinical data. The RNA sequencing (RNA-seq) dataset of UM and corresponding clinical follow-up information were downloaded from the publicly available TCGA database. This dataset was derived from the tissue samples from 80 adult patients. What’s more, 80 samples were randomly divided into dataset 1 (
Preliminary screening of mRNAs. The expression value of mRNAs was downloaded as described above. Next, we firstly screened the mRNAs with distinctive expressions among different patients of primary uveal melanoma. When the median and variance expression level of gene in every sample was more than 20% of the total median and variance expressions of all genes, the gene is considered to be a universally changed mRNA and was selected [15].
Identification of survival-related genes. In the TCGA dataset, the relationship between abundantly selected mRNA and the total survival time of patients was analyzed. Survival package in R software was used to univariable cox regression analysis. mRNAs with expressing significance
Gene Function Analysis. The function of significant survival-related genes were performed by gene ontology (GO) functional enrichment analyses and Kyoto encyclopedia of genes and genomes (KEGG) pathway. From the biological basis of an integrated knowledge and analysis tools, biological meaning of large gene or protein lists can be systematically extracted.
Identification of prognostic model relating to UM. The survival-related genes in primary selection are not suitable for clinical diagnosis. Therefore, a robust likelihood-based survival modeling approach was used to select the gene signature. We constructed a prognostic model by using the “rbsurv” package in R language.
Multivariate analysis and validation. To figure out how the multi-gene-based prognostic signature impacting the prognosis of patient samples, unsupervised hierarchical clustering analysis and multivariate survival analysis were performed on the prognostic signature by using “survival” and “survivalROC” package in R Language. The survival curves of Kaplan-Meier were drawn and the differences among groups were compared by log-rank tests. In addition, in order to assess the specificity and sensitivity of gene signature, ROC (Receiver operating characteristic) curves for predicting 5-year OS were drawn and AUC (area under the curve) values were generated [17]. Besides, in order to demonstrate the reliability of the results, the expression value of six genes was compared in 80 healthy adipose tissue samples obtained from GTEx (Genotype-Tissue Expression) database and further validated in internal datasets and outside datasets (GSE42656 and GSE84976).
Results
Preliminary screening of mRNAs. As a result, a total of 15,187 generally changed mRNAs expression values were obtained from 80 patient samples with UM after these steps. The clinicopathological characteristics of internal dataset 1 (
Clinicopathological characteristics of Datasets 1 and 2 samples
Clinicopathological characteristics of Datasets 1 and 2 samples
Identification of survival-related genes. Then we used univariate Cox regression analysis to evaluate associations between generally changed mRNAs and OS in the TCGA dataset. Totally, 2,010 survival-related mRNAs were screened out of 15,187 mRNAs. The top 20 significantly changed mRNAs were listed in Table 2.
Top 20 mRNAs significantly associated with the survival time of patients in the TCGA-UVM dataset (
Gene Function Analysis. We performed GO and KEGG functional pathway enrichment analysis in 2,010 survival-related genes. The results of GO analysis showed that the survival-related genes were significantly enriched in 18 biological pathways (listed in Table 3) related to mRNA processing, RNA splicing, mRNA transport and so on. KEGG analysis results showed that survival-related genes were significantly enriched in 6 pathways, which included Spliceosome pathway, RNA degradation pathway, Ubiquitin mediated proteolysis and so on (Fig. 1).
The functional pathway enrichment analysis of survival-related genes. (A) The GO terms. (B) The KEGG terms.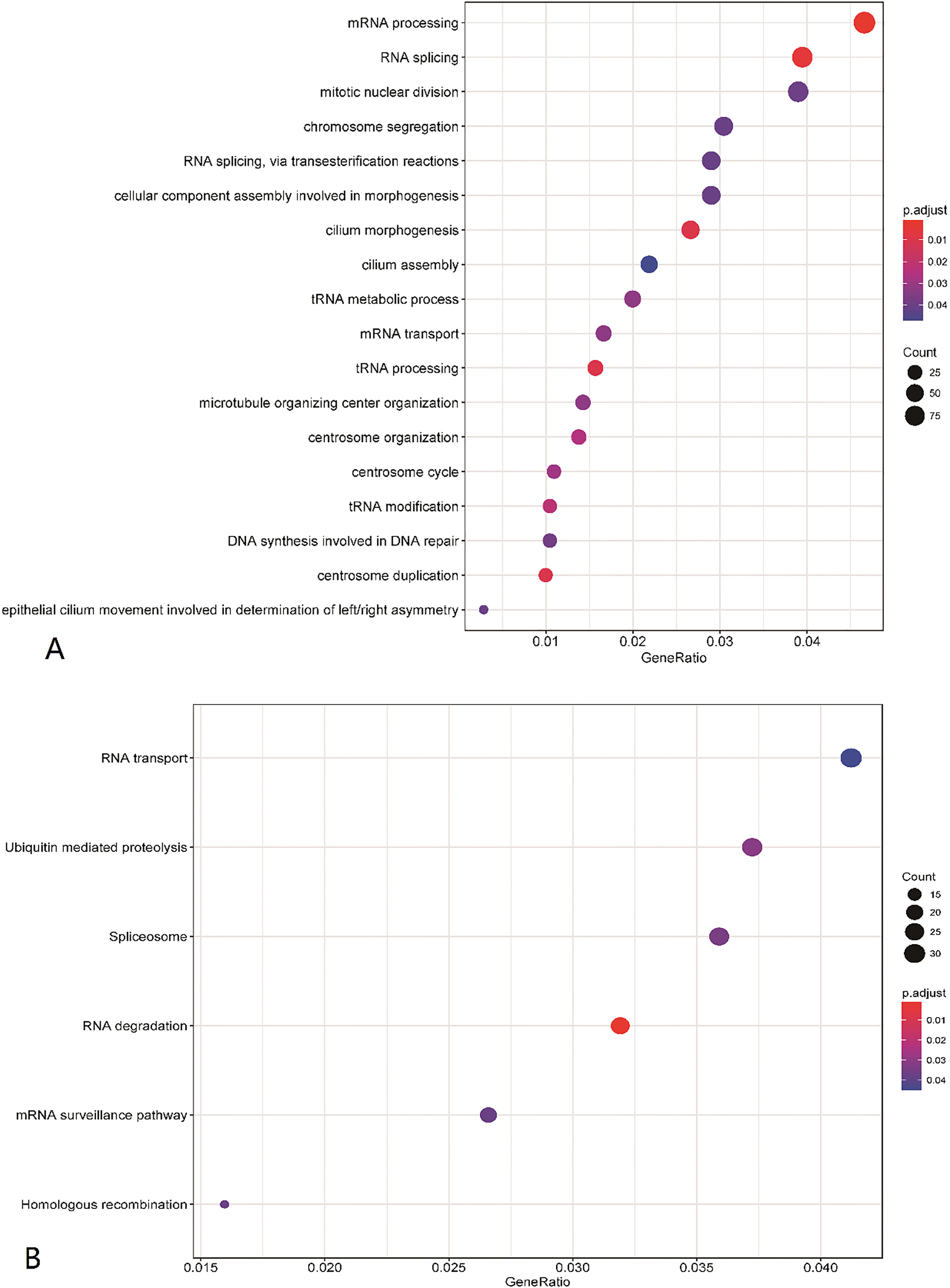
The GO and KEGG enrichment analysis of survival-related genes
Prognostic model relating to UM. Robust likelihood-based survival model approach was used to further identify robust gene combination. A series of gene models are generated by forward selection, and then the optimal model is selected by using the criterion of minimal AIC (Table 4). Finally, six of the 2,010 candidate survival-related genes were selected as signature genes to build the risk signature system that can optimally predict the OS of patients with UM. The risk system reckons a risk score for each patient. Applying the median cut-off value of the risk scores. UM patients were divided into high-risk and low-risk groups and then assessed by Kaplan-Meier method. Then, the Kaplan-Meier survival analysis of each six genes including SH2D3A, TMEM201, LZTS1, CREG1, NIPA1 and HIST1H4E were shown in Fig. 2. The ROC analysis was also used for estimating the prediction power of individual genes (Fig. 3A). Furthermore, the independent samples t-test indicated that the expression of six genes was markedly different in the tumor and normal tissues (Fig. 3B).
Prognosis related mRNA signature screened using forward selection in the TCGA-UVM dataset
Kaplan-Meier survival analysis for each six genes prognostic signature of Uveal melanoma (UM). Their expression levels were classified 2 groups as “low” and “high” .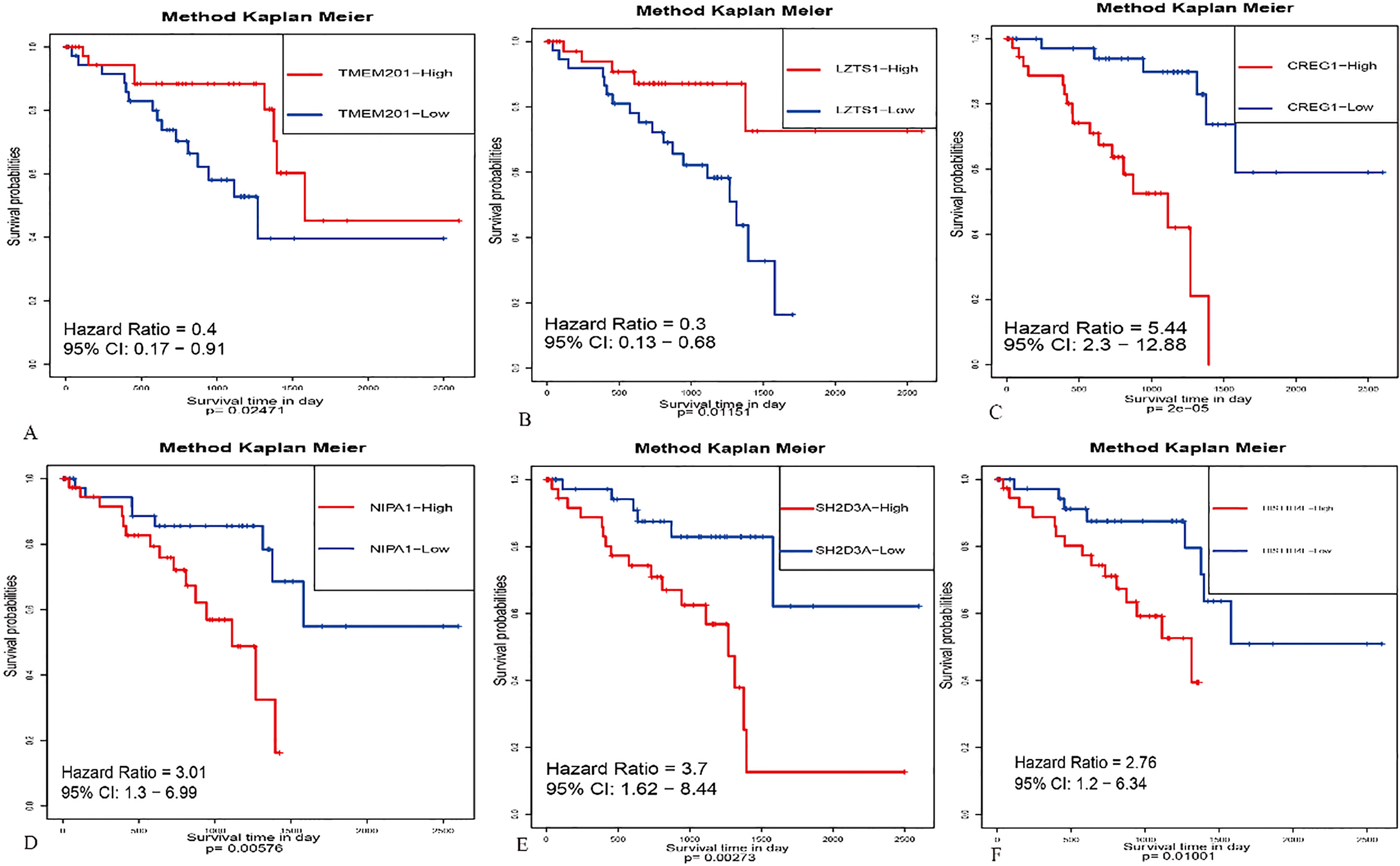
(A): The prediction power of individual genes for predicting survival of Uveal melanoma (UM). The ROC analysis was used for the discrimination between live and death cases. (B): The expression patterns of six genes in UM and healthy normal tissue (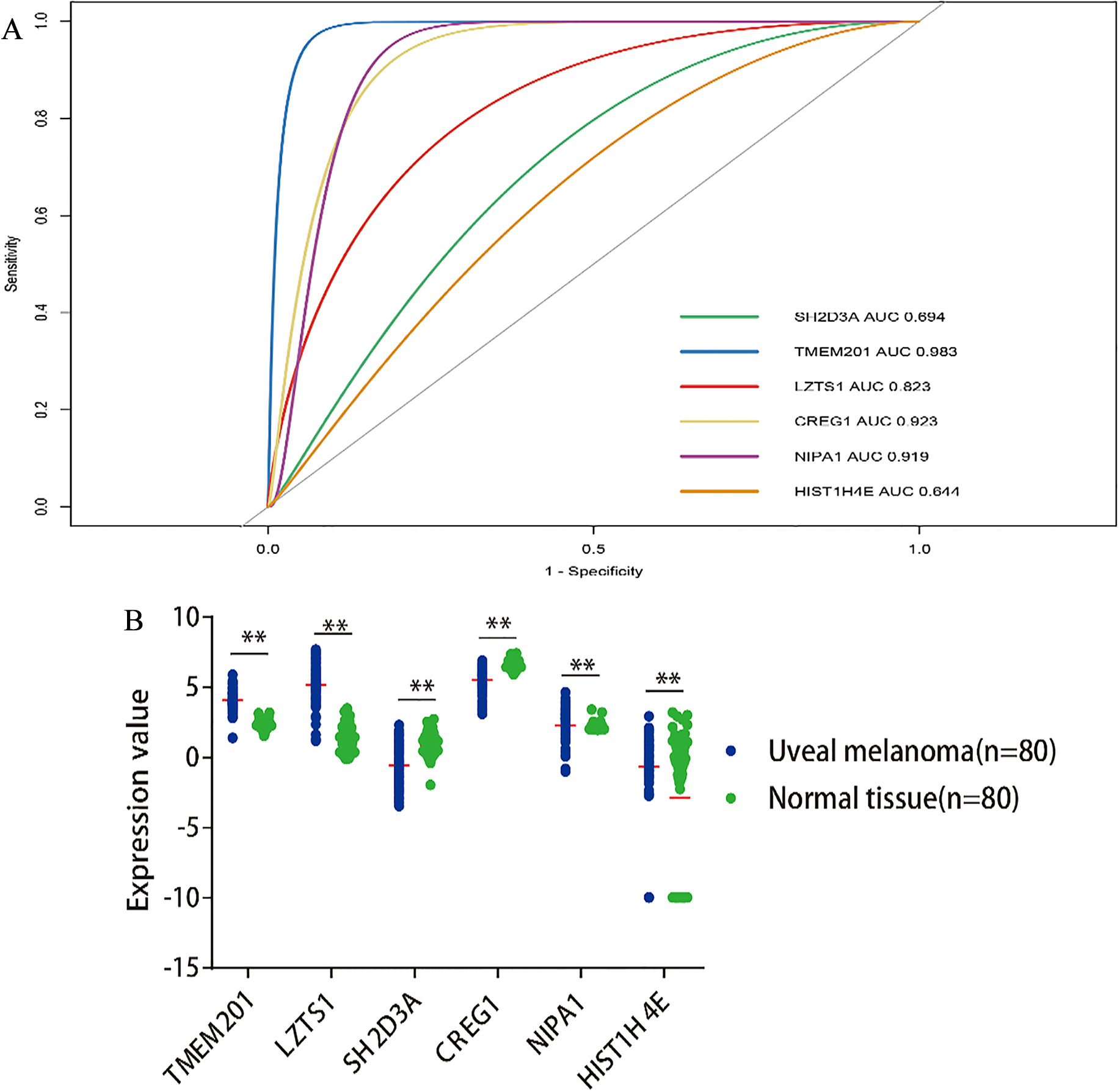
Multivariate analysis and validation. With the six-gene model, unsupervised hierarchical clustering analysis was performed in the dataset of UM, and the patients was classified into two sub-classes: Cluster 1 and Cluster 2 (Fig. 4A). Kaplan–Meier curves for two sub-classes indicated that patients in different clusters have a significant difference in OS (Fig. 4B). Therefore, this six-gene model may have important application in predicting the prognosis of UM. Moreover, we then calculated the six-gene model risk score for each patient in the TCGA and GEO datasets and ranked them according to the risk score (Fig. 5 to 7). Thus, patients were divided into two subgroups. According to their risk score in TCGA dataset and GEO datasets based on their median values and tested the significance of differences of OS between subgroups through log-rank test. The six-gene model in both internal TCGA dataset and external GEO datasets could significantly and robustly predict UM survival (shown in Fig. 8A, C, E and G). To evaluate the sensitivity of the four datasets included two internal datasets and two outside datasets, we predicted OS through the nearest neighbor method for ROC curves of censored survival value. As a result, they could effectively predict 5-year OS (shown in Fig. 6B, D, F and H).
Identification of optimal gene signature for OS prediction. (A) The Six-gene prognostic model in 80 samples were shown in a heat map (The green and the red colors represent lower and higher expression value, respectively). Unsupervised hierarchical clustering analysis was applied, which divided patients into two clusters. (B) Kaplan-Meier curves for patients in different clusters. (C) The multivariate survival analysis of the six genes. (D) The AUC curves of six genes in TCGA-UVM dataset.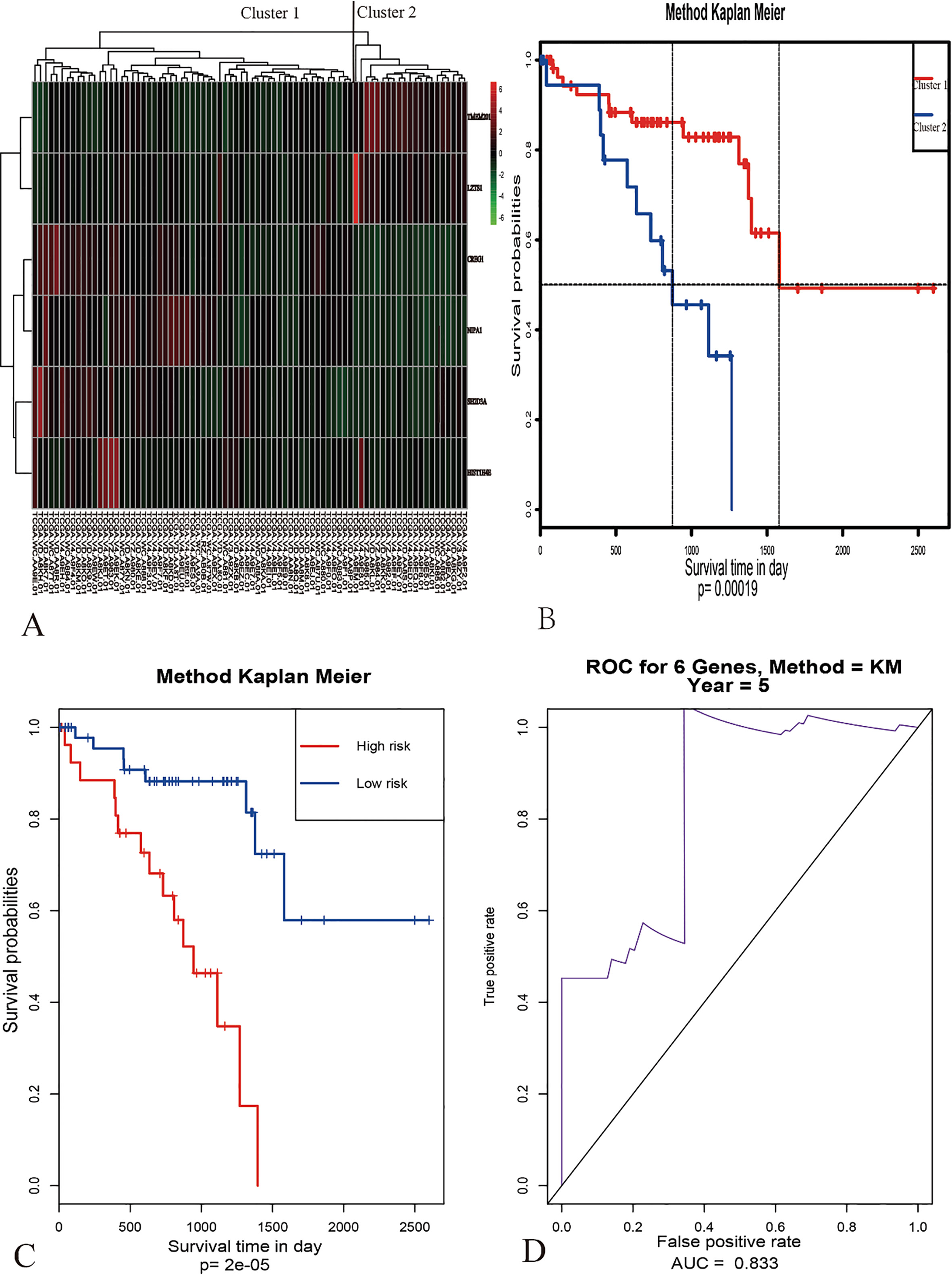
Risk score analysis of the TCGA-UVM dataset. The distribution of 6-gene based risk core, patients’ survival and gene expression signature were analysed in the TCGA-UVM dataset (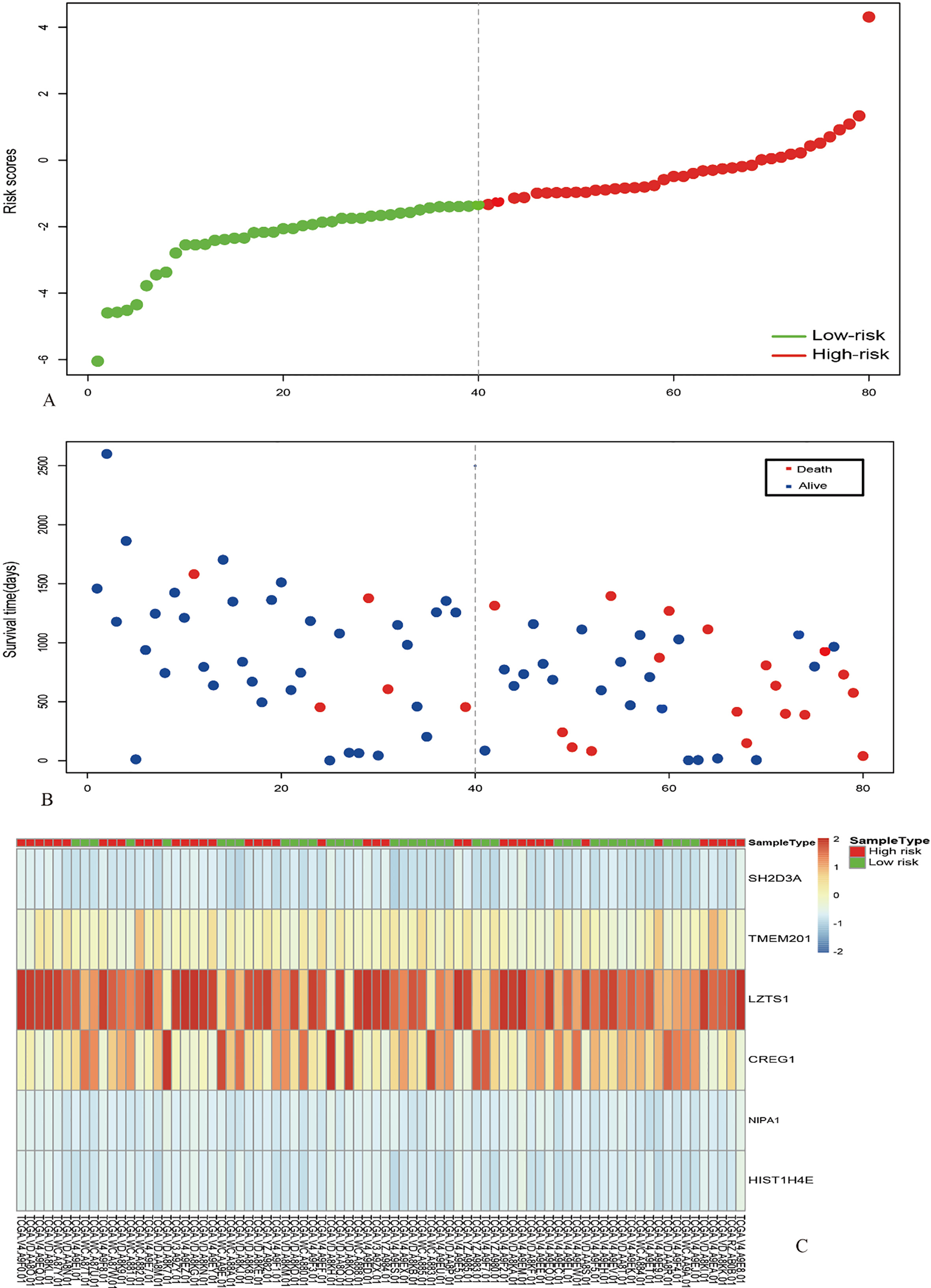
Risk score analysis of the GSE42656 dataset. The distribution of 6-gene based risk core, patients’ survival and gene expression signature were analysed in the GSE42656 dataset (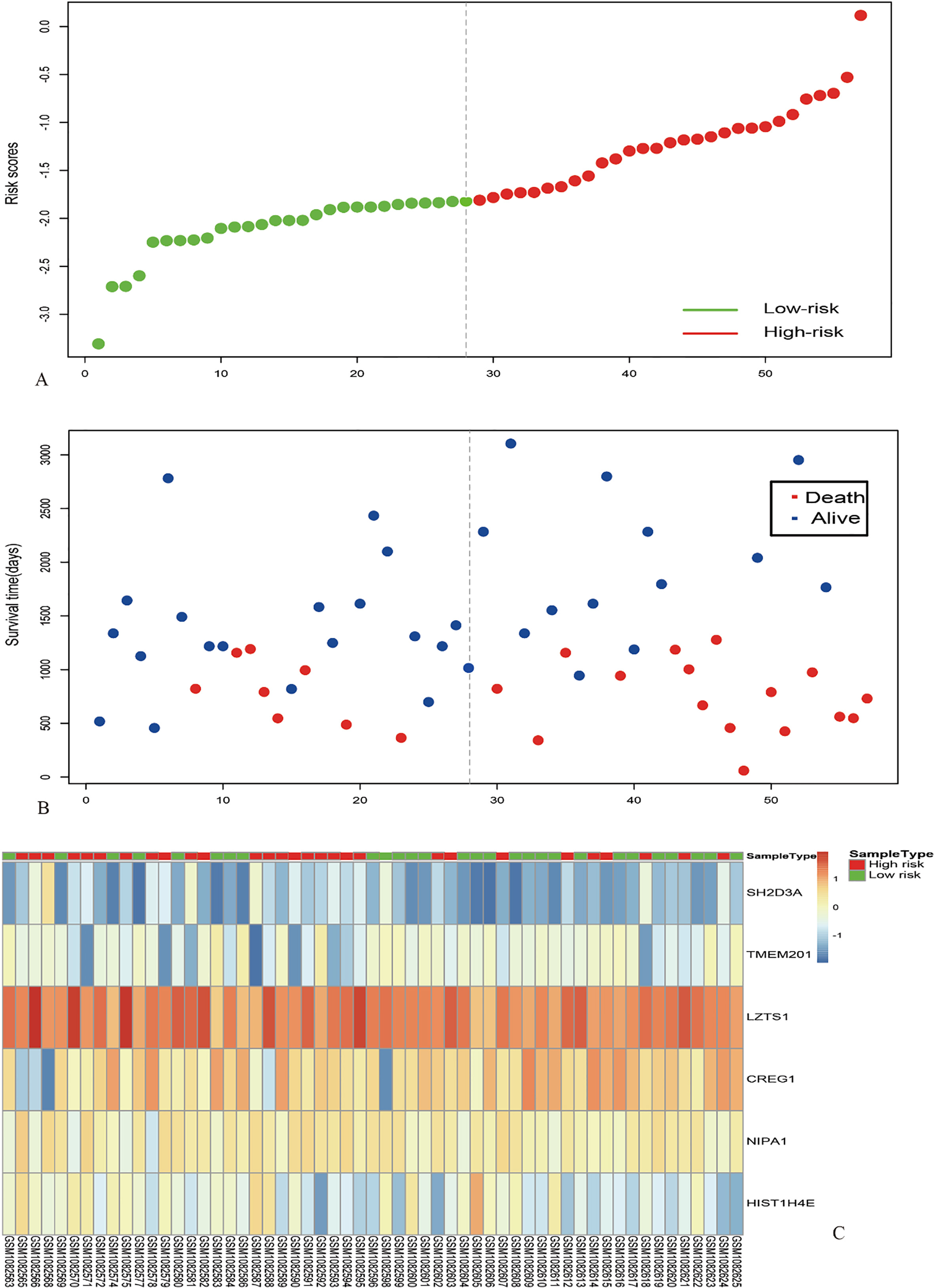
Uveal melanoma is the most common malignant ocular tumor in adults, with a mortality rate of about half of the affected patients. Although most of patients with uveal melanoma can be detected earlier for vision loss, dark spot, diopter change and visual field defect and even the best treatments have been accepted to keep from local recurrence of primary tumors, the 5-year survival rate still poor [18, 19]. It has been a problem not only in the early diagnosis but also in the prognostic evaluation. Therefore, is important to clarify the survival events of Uveal melanoma [20]. Previous evidence suggested that the abnormal expression of genes in UM were strongly associated with prognosis and can be considered as a potential prognostic factor, such as cyclin D1, p53, and MDM2 protein [21]. However, some researchers thought that the prediction accuracy of these molecules was not sufficient because they do not take into account on the simultaneous variation of multiple genes [22]. Thus, in this study, we identified 2,010 survival-related genes that are associated with survival of patients with UM. GO functional enrichment analysis showed that this survival-related genes were significantly enriched in biological pathways related to mRNA processing, RNA splicing, mRNA transport, DNA synthesis involved in DNA repair and cellular component assemble all which indicated that these pathways mainly associated with genetic material assemble processes. Valuable studies have shown that mutations in genes encoding proteins involved in RNA splicing, DNA repair, and cell component assembly occur in a variety of tumor types, including myelodysplastic syndrome, chronic lymphoblastic leukemia, and uveal Melanoma [23, 24, 25, 26]. The KEGG functional enrichment analysis showed that this survival-related genes were commonly enriched in Spliceosome, RNA degradation and Ubiquitin mediated proteolysis. Spliceosome as we known is in charge of the post-processing of primary transcripts to form mature messenger RNA species. In some solid tumors, including uveal malignant melanoma, lung adenocarcinoma and estrogen receptor positive breast cancer, high frequency splice mutations are largely reported. Splicing mutations are commonly found in various malignant tumors and may play an important role in differentiating malignant phenotypes [27, 28]. Furthermore, Ubiquitination as we known is an important post-translational protein modification which regulates a large number of important cellular processes [29, 30]. Therefore, we speculated that this survival-related genes were take most important role in the prognosis of uveal melanoma.
Risk score analysis of the GSE84976 dataset. The distribution of 6-gene based risk core, patients’ survival and gene expression signature were analysed in the GSE84976 dataset (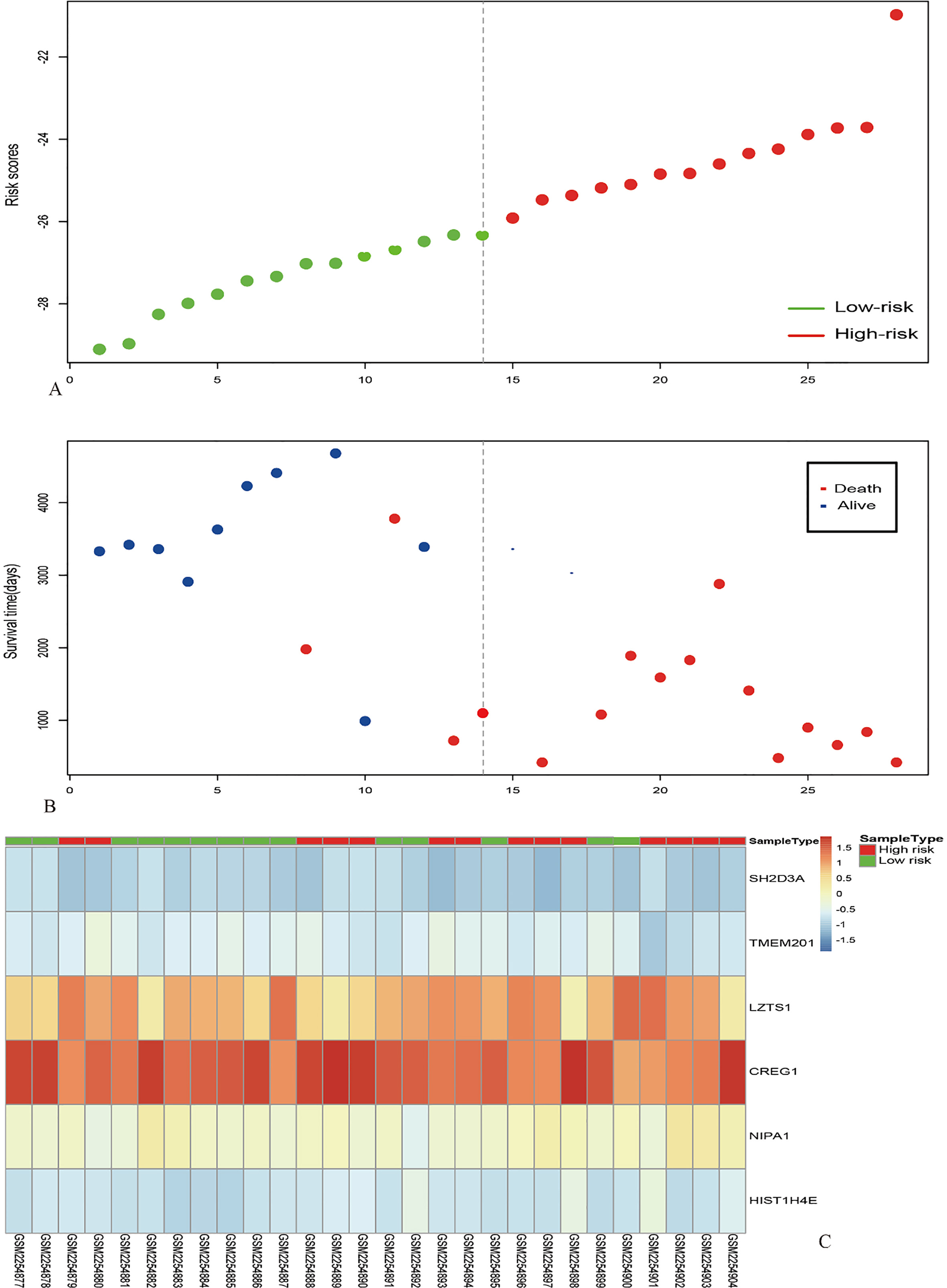
The AUC curves of 6 genes in multivariate survival analysis. (A) and (C) Kaplan-Meier survival analysis of the six genes in Dataset 1 and Dataset 2; (B) and (D) The AUC curves in Dataset 1 and Dataset 2 of TCGA-UVM dataset. (D) The AUC curves in gene expression of TCGA-UVM dataset. (E) Kaplan-Meier survival analysis in GSE42656 dataset. (F) The AUC curves in GSE42656 dataset. (G) Kaplan-Meier survival analysis in GSE84976 dataset. (H) The AUC curves in GSE84976 dataset.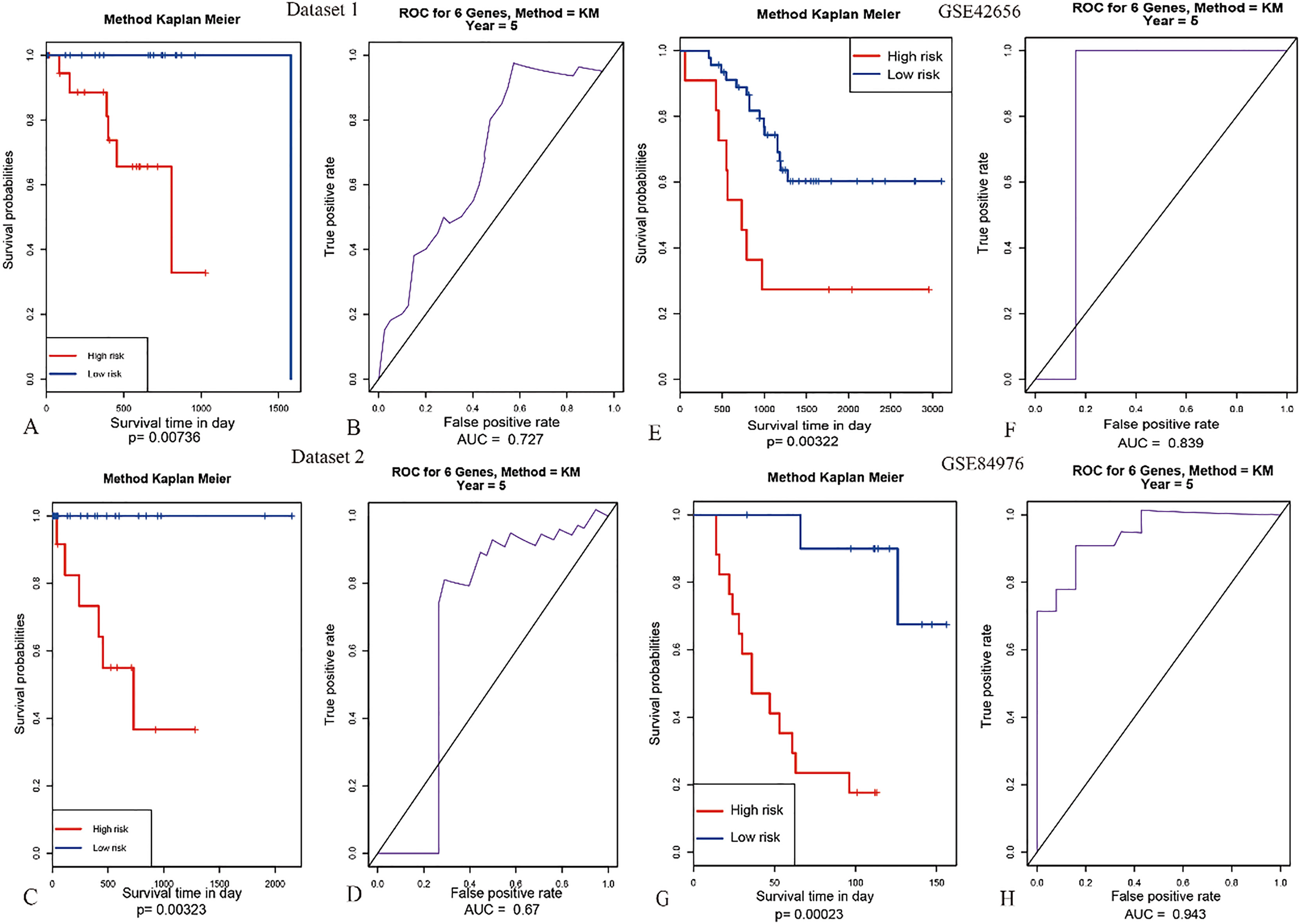
The number of survival-related genes in primary selection are large and complex is not suitable for clinical diagnosis [31, 32]. Therefore, we identified six genes that are associated with survival of patients with UM, including SH2D3A, TMEM201, LZTS1, CREG1, NIPA1 and HIST1H4E. Among the six genes, we found that high expression of SH2D3A, CREG1, NIPA1 and HIST1H4E and low expression of TMEM201 and LZTS1 were associated with significantly shorter overall survival in Uveal melanoma. Some have been reported to express in cancer or other diseases. For example, LZTS1 (leucine zipper tumor suppressor) is a tumor suppressor gene located on chromosome 8p22, which is often deleted in many human malignancies [33, 34]. Wei Zhou et al reported that LZTS1 plays a potential tumor suppressor role in colorectal cancer progression and represents a valuable clinical prognostic marker [35]. CREG1(Cellular repressor of E1A-stimulated genes) is a glycoprotein which suppresses oncogene E1A transcription and cellular transformation, which have demonstrated that CREG1 can promotes angiogenesis and neovascularization [36]. Angiogenesis has long been considered as an important factor for tumorigenesis. But for SH2D3A, TMEM201, NIPA1 and HIST1H4E, the function associated tumorigenesis remains unknown. Remarkably, the expression of these genes was markedly different expressed in the tumor and normal tissues, which suggests that these genes actively involved in tumorigenesis and might be novel oncogenes or tumor suppressor genes, and that their functions need further investigation.
At first, gene expression of the six genes prognostic signature suggested that the family of UMs in TCGA can at least divided into two sub-classes and have association of UM subtype with a trend toward longer survival among patients. Moreover, the six genes prognostic signature was validated in two internal datasets and another two independent patient sets on different platforms. In fact, by ROC curve analysis, this six genes prognostic signature can be used to predict OS in patients with UM from three datasets (TCGA-UVM, GSE42656 and GSE84976) with an AUC of 0.833, 0.839 and 0.943 respectively. Multivariate analysis showed that the six-gene prognostic signature was an independent prognostic factors for 5 years OS in UM patients [37] (Fig. 8). Compared with other studies based on different prognostic models, this study obtained a signature with only six genes to successfully predict the OS in UM patients. It is a great significance to establish a PCR detection method for clinical application [38, 39].
Although we identified a prognostic signature and pathways for prognostic of uveal melanoma, there are some limitations to our study. The study was carried out based on bioinformatics methods and the conclusions have not been proved by experiments. Additionally, the sample size in our study was limited. Hence, large sample size of patients is warranted to further explore the molecular mechanisms of UM.
In summary, we identified a six-gene prognostic signature for UM from TCGA dataset and have validated in GEO dataset, which should be helpful for UM early diagnosis and might be regarded as new promising biomarkers for UM prognosis and treatment.
Footnotes
Acknowledgments
This work was supported by The National Key R&D program of China (2018YFC1106000).
Conflict of interest
All authors declare no conflict of interest.