
Abstract
As a result of metastasis and high recurrence, ovarian carcinoma (OC) is one of the most frequent gynecological carcinomas affecting women up to now. In spite of advances in OC treatments, the molecular mechanisms underlying OC progression are still needed to be deeply understood. MicroRNAs (miRNAs) with aberrant expressions are widely known to regulate target genes so as to mediate diverse biological activities of tumor cells. In the present study, we inspected the expression profile and latent mechanism of miR-3666 in OC. First of all, our research revealed the down-regulated miR-3666 in OC cells. Furthermore, miR-3666 up-regulation could repress cell proliferation and migration as well as induce cell apoptosis in OC. In addition, we unmasked that miR-3666 targeted STAT3 (signal transducer and activator of transcription 3) and further down-regulated STAT3 expression. Moreover, adenylate kinase 4 (AK4) was transcriptionally enhanced by STAT3, and then miR-3666 restrained AK4 expression by mediating STAT3. In the end, rescue experiments depicted that miR-3666 suppressed the development of OC via STAT3-mediated AK4. We uncovered that miR-3666 inhibited the tumorigenesis and even development of OC via suppressing STAT3/AK4 axis, offering a novel biomarker and therapeutic target for OC.
Introduction
Ovarian carcinoma (OC) is one dominating malignant cancer in the female genital system, ranking as the highest mortality for gynecologic cancers [1]. It generally comes with a risk of poor prognosis, especially in older patients [2]. Although researchers have excavated different therapies for OC, the outcome of OC patients is still disappointing because of drug resistance and undiscovered pathogenesis [3].
Though the molecular mechanism in OC cells is not thoroughly investigated, its high dependence on the abnormal expression of cellular genes affecting neoplasia potential has been validated by numerous reports [4, 5]. For example, long noncoding RNA (lncRNA) SNHG15 predicts poor prognosis in patients with epithelial OC [6]. LncRNA CASC9 sponges miR-758-3p to promote LIN7A expression for enhancing the malignancy in OC [7]. MiR-381 inhibits cell migration and invasion in gastric cancer through down-regulating SOX4 [8].
In this regard, microRNAs (miRNAs) are one type of short noncoding RNAs comprising 18 to 25 nucleotides with conserved sequences [9]. It has been reported that miRNAs can lower the expression of target mRNAs by bringing about mRNA degradation or restraint of translation via binding to the 3’-UTR area, therefore impacting basal biological processes in tumors including OC [10, 11]. For instance, miR-27a serves as a tumor-promoter in OC through targeting FOXO1 [12]. MiR-552 aggravates ovarian carcinogenesis via modulating PTEN signaling [13]. Nevertheless, most miRNAs have not been studied in OC.
Increasing researches have emerged the low-grade expressed miR-3666 as a tumor-suppressor in multiple cancers. For example, miR-3666 regulates cell proliferation in thyroid carcinoma via MET [14]. MiR-3666-caused suppression of SIRT7 inhibits cell growth in non-small cell lung cancer [15]. Pituitary tumor-transforming gene 1 promotes metastasis in cervical cancer via miR-3666-regulated ZEB1 [16]. Still and all, the role and molecular mechanism underlying miR-3666 in OC have not been figured out till now.
Compelling evidence has established that the transcription factor signal transducer and activator of transcription 3 (STAT3) is active in a number of human cancers, and its aberrant activity influences the development of tumors through boosting cell proliferation and migration, suppressing cell apoptosis and preventing against immune surveillance [17, 18]. Previously, STAT3 has been unveiled to be necessary for OC development [19], and targeting STAT3 has been proposed as a promising therapeutic method for OC [20]. Further, STAT3 is suggested to function as a transcription factor to modulate gene expression at transcriptional level in carcinomas. For example, LINC00165 activated by STAT3 develops carcinogenic properties via binding to Polycomb Repressive Complex 2 to enhance EMT in gastric cancer [21]. Such role of STAT3 has also been reported in OC. Jin et al. found that STAT3 could activate miR-216a to facilitate OC cell proliferation and chemo-resistance [22]. STAT3 elevates MMP9 to enhance the invasiveness of OC cells [23]. However, many about the upstream and downstream of STAT3 have not been explored in OC.
Adenylate kinase 4 (AK4) is a highly-recognized oncogene in diverse malignancies. For instances, AK4 contributes to cell proliferation and invasion in bladder cancer [24] and breast cancer [25]; AK4 affects drug sensitivity in HeLa and A549 cells [26]; AK4 has an important role in accelerating metastasis in lung cancer [27, 28]. However, whether AK4 is involved in OC development and how it is regulated in OC are still veiled.
In the present study, we inquired the expression profile and potential mechanism of miR-3666 in OC. As a result, we discovered that overexpression of miR-3666 repressed OC progression. Besides, miR-3666 could down-regulate AK4 expression by targeting STAT3. In the end, rescue experiments displayed that miR-3666 suppressed the development of OC via silencing STAT3-activated AK4.
Materials and methods
Cell culture
OC cell lines (ES2, SKOV-3, CAOV-3 and HO8910) and the normal fallopian tube epithelial cell line (FTE187) bought from American Type Culture Collection were kept in DMEM medium with 100
Cell transfection
For STAT3 or AK4 overexpression, STAT3 or AK4 mRNA sequence was compounded and cloned into the pcDNA3.1 vector (Invitrogen), and the empty pcDNA3.1 vector was used as the negative control. Meanwhile, miR-3666 mimic and negative control mimic (NC-mimic) were purchased from Ribobio. These plasmids were transfected into OC cells utilizing Lipofectamine 2000 (Invitrogen) as instructed by the manufacturer.
Quantitative real-time polymerase chain reaction (RT-qPCR) analysis
Total RNA from OC cells was harvested with TRIzol reagent (Life Technologies) under the manufacturer’s protocol. PrimeScript RT reagent Kit (TaKaRa, Japan) was employed to implement reverse transcription and SYBR Green Real-Time PCR Master Mix (Thermo Fisher Scientific, USA) was applied for qPCR on ABI 7500 PCR System (Applied Biosystems, USA). The internal controls were U6 for miR-3666 and GAPDH for the others (including STAT3 and AK4). The prepared primer sequences were exhibited as follow: miR-3666 (forward: 5’-CAGTGCAAGTGTAGATGCC-3’ and reverse: 5’-GAACATGTCTGCGTATCTC-3’), STAT3 (forward: 5’-CAGCAGCTTGACACACGGTA-3’ and reverse: 5’-AAACACCAAAGTGGCATGTGA-3’), AK4 (forward: 5’-TGGATTCACCCTCCTAGCGGAA-3’ and reverse: 5’-CTGTCTTAGCCTGGCAGCAACT-3’), U6 (forward: 5’-CTCGCTTCGGCAGCACA-3’ and reverse: 5’-AACGCTTCACGAATTTGCGT-3’) and GAPDH (forward: 5’-ACAGTCAGCCGCATCTTCT-3’ and reverse: 5’-GACAAGCTTCCCGTTCTCAG-3’).
Cell counting Kit-8 (CCK-8) assay
Cell viability was tested through CCK-8 assay. In brief, 4
Colony formation assay
The collected ES2 and CAOV3 cells (500 cells per well) were seeded into 6-well plates 48 h after transfection. Following 14 days of incubation, the resulting colonies were treated with methanol for 30 min and with 0.5% crystal violet solution for 15 min. The visible colonies were counted manually.
EdU assay
EdU assay in transfected ES2 and CAOV3 cells was achieved by using BeyoClick™ EdU Cell Proliferation Kit (Beyotime, Shanghai, China). Cell samples were rinsed with PBS in 96-well plates before culturing with EdU medium for 2 h. Following fixing for 30 min by 4% PFA, cells were stained by DAPI solution for observing cell nuclei. The inverted microscope (Olympus, Tokyo, Japan) was used for analysis as instructed by the provider.
Caspase-3 activity assay
Cell apoptosis was analyzed by caspase-3 activity assay with a commercial kit (Roche Life Science, Shanghai, China). The supernatant from the cell lysates in glacial cell lysis buffer was collected, and then protein concentration was examined. After that, protein (100
Transwell assay
The migration of OC cells was assessed with transwell chambers (8
Western blot analysis
The acquired total protein was used for separation on SDS-PAGE (10%), and then shifted onto PVDF membranes. After blocking by 5% nonfat milk, PVDF membranes were probed overnight at 4
RNA pull down assay
The miR-3666 sequence which contained wild-type or mutated STAT3 binding sites were biotin-labeled into Bio-miR-3666-WT/Mut, by using Biotin RNA Labeling Mix (Roche, Basel, Switzerland). After that, the Bio-NC or Bio-miR-3666-WT/Mut probes were mixed with cell lysates and magnetic beads overnight. After washing, RNA in the mixture pulled down was evaluated by RT-qPCR.
Luciferase reporter assay
To detect the interplay between STAT3 and miR-3666, the fragments of STAT3 3’UTR containing the potential miR-3666-binding sites were enlarged by PCR and cloned into the pmirGLO dual-luciferase miRNA target vectors, forming the reporter vectors of STAT3-wild-type (STAT3-WT). Similarly, the sequence of STAT3 3’UTR with mutant miR-3666-binding sites were obtained and inserted into pmirGLO vectors, forming the reporter vectors of STAT3-mutant (STAT3-Mut). To detect the combination of STAT3 protein with AK4 promoter, AK4 promoter was also subcloned into the pGL3 reporter vectors to construct pGL3-AK4 promoter reporters. Then, with the help of Lipofectamine 300, STAT3-WT or STAT3-Mut was co-transfected with miR-3666 mimic or NC-mimic into the cells, and pGL3-AK4 promoter reporter was con-transfected into cells with pcDNA3.1 or pcDNA3.1-STAT3. 48 h later, the dual-luciferase reporter assay system (Promega, USA) was adopted to determine the luciferase activity, as instructed by the provider.
Overexpression of miR-3666 inhibited cell proliferation and migration and induced cell apoptosis in OC. (A) RT-qPCR analysis of miR-3666 expression in OC cell lines (ES2, SKOV-3, CAOV3 and HO8910) and the normal fallopian tube epithelial cell line (FTE187). (B) The overexpression efficiency of miR-3666 by miR-3666 mimic in ES2 and CAOV3 cells was tested by RT-qPCR. (C) The viability of miR-3666 mimic transfected ES2 and CAOV3 cells was examined through CCK-8 assay. (D-E) The proliferation of miR-3666 mimic transfected ES2 and CAOV3 cells was detected by colony formation and EdU assays. (F) The apoptosis rate of ES2 and CAOV3 cells transfected with miR-3666 mimic was measured by caspase-3 activity detection. (G) Transwell assay was used to assess the migration of ES2 and CAOV3 cells in two groups. 
The EZ Magna RNA immunoprecipitation Kit (Millipore, USA) was employed for RIP experiments referring to the guidelines of the manufacturer. Briefly, the lysates from ES2 and CAOV3 cells in RIP lysis buffer were subjected to the incubation of magnetic beads with anti-Ago2 or anti-IgG (Millipore) at 4
Chromosome immunoprecipitation (ChIP)
4% formaldehyde was applied for fixing the cross-linking in cells at room temperature for 10 min, and then 0.125 M glycine was added for stopping the cross-linking reaction, followed by shear of DNA into an average scale of approximately 500 bp with the Vibra cell sonicator (Sonics and Materials inc; Newtown, CT, USA). Then the sonicated mixture was centrifuged at 14,000 rpm for one quarter at 4
Statistical analysis
Data processed by SPSS 18.0 statistical software package were shown as mean
MiR-3666 directly targeted and further down-regulated STAT3. (A) The feasible binding sequence between miR-3666 and STAT3 was obtained from TargetScan. The mutant sequence of STAT3-Mut with the predicted miR-3666 binding sites mutated was constructed. (B) RNA pull down assay measured the enrichment of STAT3 in Bio-miR-3666-WT, Bio-miR-3666-Mut and Bio-NC groups. (C-D) Luciferase reporter assay and RIP assay were performed to confirm the interaction between miR-3666 and STAT3. (E) STAT3 mRNA and protein expressions under miR-3666 promotion were determined by RT-qPCR and western blot. 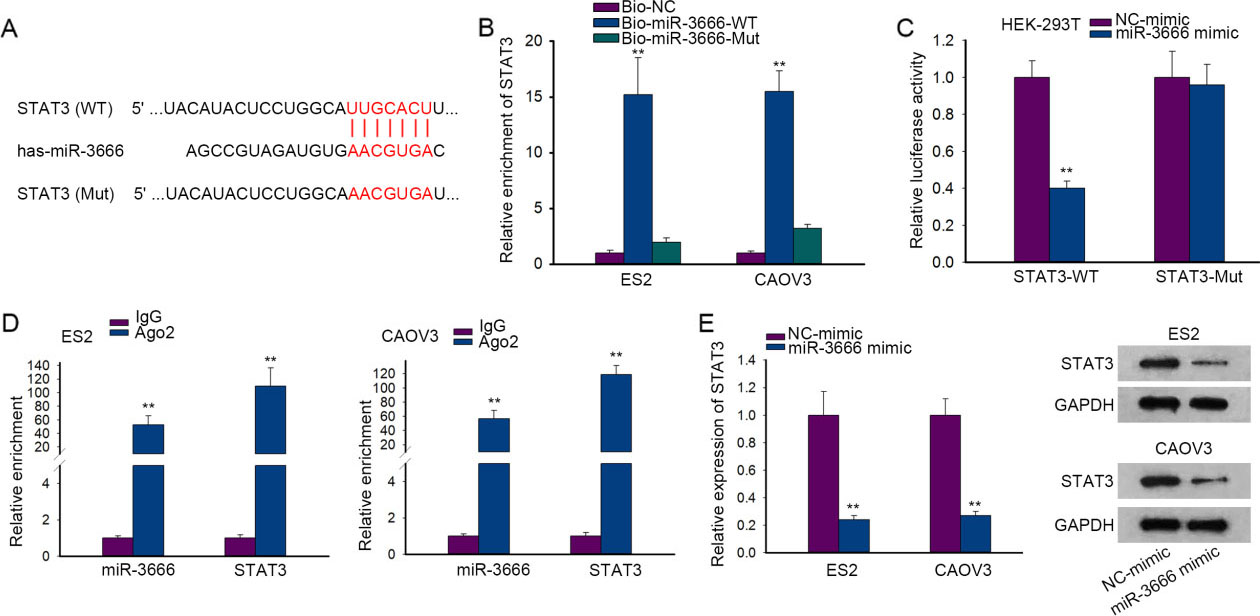
Overexpression of miR-3666 inhibited cell proliferation and migration in OC
To inspect whether miR-3666 took part in the tumorigenesis and development of OC, we examined the expression level of miR-3666 in OC cell lines (ES2, SKOV-3, CAOV-3 and HO8910) and the normal fallopian tube epithelial cell line (FTE187). Results showed that miR-3666 expression was relatively lower in OC cell lines than that in FTE187 cells (Fig. 1A). Among the cultured cell lines, ES2 and CAOV3 cells exhibited the lowest expression of miR-3666. Based on this, we chose ES2 and CAOV3 cells for later assays. It was validated that the expression level of miR-3666 was significantly increased by miR-3666 mimic (Fig. 1B). Then we explored the biological role of miR-3666 in OC through gain-of-function assays. According to the results of CCK-8 experiment, cell viability was inhibited by miR-3666 overexpression compared with the control group (Fig. 1C). Further, data from colony formation and EdU assays validated that miR-3666 overexpression impaired the proliferation ability of OC cells (Fig. 1D–E). Conversely, we discovered that the caspase-3 activity was increased under miR-3666 upregulation (Fig. 1F), hinting that miR-3666 facilitated OC cell apoptosis. Moreover, transwell assay data showed that cell migration was repressed in response to the overexpression of miR-3666 (Fig. 1G). To sum up, miR-3666 was down-regulated in OC cells and miR-3666 overexpression hindered the development of OC.
MiR-3666 directly targeted and further down-regulated STAT3
Emerging evidence has reported that miRNAs modulate gene expression by interacting with 3’UTR of target mRNAs [11]. In this case, we were then interested in the downstream target of miR-3666 in OC. From TargetScan, we found the probable interaction between miR-3666 and STAT3 (signal transducer and activator of transcription 3) and obtained the binding sequence between them (Fig. 2A). Subsequently, the outcomes of RNA pull down assay testified the obvious enrichment of STAT3 in Bio-miR-3666-WT group rather Bio-miR-3666-Mut or Bio-NC group (Fig. 2B). Further, luciferase reporter assay data proved that the up-regulation of miR-3666 merely reduced the luciferase activity of STAT3-WT (Fig. 2C). Besides, RIP assay results uncovered that miR-3666 and STAT3 were both enriched in the complex precipitated by anti-Ago2 (Fig. 2D), validating the combination between miR-3666 and STAT3 in RNA-induced silencing complexes (RISCs). Importantly, it was verified by RT-qPCR and western blot that STST3 mRNA and protein expressions were both markedly decreased in OC cells after miR-3666 overexpression (Fig. 2E). Taken together, miR-3666 directly targeted and down-regulated STAT3 in OC cells.
MiR-3666 targeted STAT3 to down-regulate AK4 at transcriptional level. (A) The potential transcriptional regulation of STAT3 on AK4 as browsed on UCSC (
Transcription factors like STAT3 can transcriptionally modulate gene expression [29]. Here, we discovered that adenylate Kinase 4 (AK4) was under the transcriptional regulation of STAT3 according to UCSC Genome Browser (
Up-regulation of AK4 rescued the inhibition of miR-3666 overexpression on OC progression. ES2 and CAOV3 cells were individually co-transfected with NC-mimic 
To testify the regulation mechanism of miR-3666 in OC, we conducted rescue experiments. The overexpression efficacy of AK4 by pcDNA3.1-AK4 was assessed by RT-qPCR (Fig. 4A). Then, the results of CCK-8, colony formation and EdU experiments showed that cell proliferation was restrained by miR-3666 overexpression but recovered partly by AK4 up-regulation (Fig. 4B–D). Also, we found that overexpression of miR-3666 reinforced the caspase-3 activity, which was then lowered partially owing to the up-regulation of AK4 (Fig. 4E). Likewise, transwell assay data represented that the inhibited cell migration caused by overexpressed miR-3666 was reversed after up-regulating AK4 (Fig. 4F). In general, miR-3666 down-regulated AK4 to inhibit OC progression.
Discussion
Ovarian carcinoma (OC) is the staple cause of gynecological cancer-associated mortality in the world since OC patients generally diagnosed at advanced stages [30]. As a result of the complexity in the molecular mechanism of OC progression, the therapy outcomes of OC patients have largely lagged behind. Therefore, the searching of latent valid biomarkers is the first requisite for developing early diagnosis method and clinical treatment of OC patients [31].
MicroRNAs (miRNAs) have been acknowledged to modulate gene expression so as to exert oncogenic or tumor-suppressive functions in human carcinomas [32, 33, 34]. Several reports indicate that miR-3666 acts as a tumor-suppressor in different cancer types, such as thyroid cancer [14], lung cancer [15], cervical cancer [16] and colorectal cancer [35]. Consistently, our work demonstrated that miR-3666 was down-regulated in OC and its up-regulation inhibited OC cell proliferation and migration. Of note, it was the first time to probe the role of miR-3666 in OC.
From TargetScan, we gained the target genes of miR-3666, among which Signal Transducer and Activator of Transcription 3 (STAT3) was the option in this paper. STAT3, a transcription activator, is known as an oncogene in a diverse range of tumors [36, 37]. For instance, STAT3 boosts the constitutive activity of AGC kinases in melanoma via trans-activating PDK1 [29]. The aspirin-induced lncRNA OLA1P2 blocks the homodimer formation of phosphorylated STAT3 in colorectal cancer [38]. Interestingly, researches have also verified the importance of STAT3 in OC [19, 20]. In the present work, we uncovered that STAT3 was indeed a target downstream of miR-3666 in OC cells.
Subsequently, we investigated the possible downstream effector of miR-3666/STAT3 pathway in OC. As is known to all, adenylate kinase-4 (AK4) is recognized as a carcinogene in lung cancer [39] and osteosarcoma [40], whereas its role in OC has not been explored. Here, we intriguingly found the transcriptional regulation of STAT3 on AK4. Further, current study also validated that miR-3666 targeted STAT3 to down-regulate AK4 in OC cells. Moreover, the results of rescue experiments presented that miR-3666 restrained OC progression through targeting AK4.
In sum, the findings in our work offered a mechanistic account for the miR-3666/STAT3/AK4 pathway in OC, replenishing the rational of miR-3666-targeted therapy in OC.
Footnotes
Acknowledgments
Thanks for all involved in this work. This work was supported by the National Natural Science Foundation of China under grants 81401054 and 81402125.