
Abstract
OBJECTIVE:
The aim of this work was to extensively explore the long non-coding RNA (lncRNA) expression profiles in acute myeloid leukemia (AML) and to propose candidate lncRNAs with predictive value for AML risk.
METHODS:
The bone marrow mononuclear cell samples from 10 AML patients and 10 age and gender matched controls were obtained and subjected to next-generation RNA sequencing. Then, the top 5 upregulated and top 5 downregulated lncRNAs in AML patients compared with controls were selected for further quantitative polymerase chain reaction (qPCR) validation in 40 AML patients and 40 age and gender matched controls. The effect of candidate lncRNA RP11-342M1.7 on proliferation and apoptosis in AML cells was further investigated.
RESULTS:
RNA sequencing observed 216 upregulated and 412 downregulated lncRNAs in AML patients compared with controls. Enrichment analyses exhibited that these differentially expressed lncRNAs were involved in neoplastic signaling pathways such as cAMP and MAPK signaling pathways. In further qPCR validation, lncRNA RP11-342M1.7 and lncRNA CDCA4P3 were upregulated, while lncRNA CES1P1, lncRNA AC008753.6 and lncRNA RP11-573G6.10 were downregulated in AML patients compared with controls. Multivariate logistic regression analysis disclosed that lncRNA RP11-342M1.7, lncRNA CES1P1 and lncRNA AC008753.6 were independent predictive factors for AML risk, and most importantly, the combination of these three lncRNAs was of remarkably good predictive value for AML risk (AUC: 0.901; 95% CI: 0.835–0.966). Besides, lncRNA RP11-342M1.7 was correlated with higher CR while lncRNA AC008753.6 and lncRNA CTD-2562J15.6 were correlated with lower CR. LncRNA RP11-342M1.7 knockdown suppressed cell proliferation by promoting cell apoptosis in AML cells.
CONCLUSIONS:
This study reveals the comprehensive lncRNAs expression profiles in AML, and proposes candidate lncRNAs that are potential biomarkers for AML risk.
Introduction
Acute myeloid leukemia (AML) as the most common myeloid leukemia in adults, is a highly heterogeneous hematological malignancy characterized by abnormal accumulation of poorly differentiated myeloid cells in the bone marrow and failure in normal hematopoiesis [1]. The incidence of AML is around 3 to 8 cases per 100,000 in adults aged 18 to 60 years, and 9 to 17 cases per 100,000 in adults aged over 65 years [2]. Although previous studies have demonstrated that the genetic background as well as environmental factors contribute to the pathogenesis of AML, the exact mechanism underlying the onset of AML is still undetermined [3]. Benefiting from whole genome sequencing, the genetic abnormities in AML have been partially revealed and contribute to the disease classification as well as development of target therapies, however, the biological intricacies of AML are far more complicated and the clinical outcomes of most of AML patients remain unsatisfactory. Therefore, the discovery of novel molecular markers might lead to better understanding of the pathogenesis of AML and explore novel therapeutic targets to improve clinical outcomes in AML patients.
Long non-coding RNAs (lncRNAs) are a novel class of non-coding RNAs more than 200 nucleotides long, which are involved in various cellular processes such as chromosomal remodeling, transcription and intracellular trafficking [4]. Over the past decade, lncRNAs emerge as the key regulators in human malignancies, and with the advances of next generation high-throughput RNA sequencing, the expression profiles and functions of lncRNAs are springing up [5]. In leukemias, a number of individual lncRNAs such as CRNDE, HOTAIRM1, DLEU1, DLEU2, LUNAR1, BLG3 and MALAT1 are reported to be implicated in leukemic progression [4]. As for in leukemias, 2560 novel lncRNAs are identified to be aberrantly expressed in T cell acute lymphoblastic leukemia cell lines (DND41, Jurkat and RPMI-8402), and over 1,300 lncRNAs and 940 mRNAs were differentially expressed in chronic lymphocytic leukemia patients with progressive disease compared to those with indolent disease [6, 7]. In AML, a subset of lncRNAs are found to be subtype-specifically enriched in AML patients, and 111 lncRNAs are correlated with gene expression signature of leukemia stem cells of AML [8, 9]. However, the functions of the vast majority of lncRNAs are still uninvestigated in AML. In order to provide valuable information for further research about lncRNAs and to study the potential of lncRNAs as biomarkers in AML, we extensively explored the lncRNA expression profiles in AML and proposed candidate lncRNAs with predictive potential for AML in this present study.
Methods
Participants
Forty AML patients admitted on Huangshan City People’s Hospital from Jan 2018 to Feb 2019 were consecutively enrolled in this study. The inclusion criteria were as follows: confirmed diagnosis of de novo AML, age above 18 years. The exclusion criteria were as follows: secondary AML or history of other solid tumor/hematological malignancies, diagnosis of acute promyelocytic leukemia, previous radiotherapy, chemotherapy or targeted therapies, severe heart dysfunction/arrhythmia/lung dysfunction/hepatic dysfunction/renal dysfunction. Meanwhile, 40 healthy donors with age and gender matched to AML patients were also enrolled as controls. This study was approved by the Ethics Review Board of Huangshan City People’s Hospital, and all participants signed the informed consents before enrollment.
Data collection
After enrollment, the following information of AML patients were collected: age, gender, white blood cell count (WBC), French-American-British classification systems (FAB) classification, cytogenetics information, mutations of internal tandem duplications in the FMS-like tyrosine kinase 3 (FLT3-ITD), isolated biallelic CCAAT/enhancer-binding protein
Treatment and assessment
Three days of an anthracycline and 7 days of cytarabine (“3
Sample acquisition
Bone marrow samples were obtained from AML patients before the initiation of any treatments and from controls after enrollment by biopsy. Then bone marrow mononuclear cells (BMMCs) were isolated by Ficoll-Hypaque density gradient centrifugation.
RNA sequencing
Ten AML patients and 10 age/gender matched controls were randomly selected from total participants, and then their BMMCs were subjected to RNA sequencing for detection of lncRNA and mRNA expres- sion profiles (RNA sequencing stage). In brief, total RNA was extracted using Trizol reagent (Invitrogen, USA) and then quality control was performed (concentration, purity and integrity). Then ribosomal RNA (rRNA) was removed using Epicentre Ribo-zero
Bioinformatic analysis
The bioinformatics analysis for RNA sequencing was performed using R software (Version 3.3.3). Genes (including lncRNA and mRNA) which were detected in 50% or above samples were included in the analysis. Principal component analysis (PCA) was performed using Stats packages. Dysregulated genes were analyzed using DeSeq2 package and showed as Volcano spot, the statistical significance was defined as
Quantitative polymerase chain reaction (qPCR)
Top 5 upregulated and top 5 downregulated lncRNAs in AML patients compared to controls in RNA sequencing stage were further included in the qPCR validation stage including lncRNA RP11- 342M1.7, lncRNA AC074363.1, lncRNA CDCA4P3, lncRNA LINC01602, lncRNA RP11-754N21.1, lncRNA CES1P1, lncRNA AC008753.6, lncRNA LINC01220, lncRNA CTD-2562J15.6 and lncRNA RP11-573G6.10. These lncRNAs were detected in BMMCs derived from 40 AML patients and 40 age and gender matched controls by qPCR. In brief, total RNA was extracted using Trizol reagent (Invitrogen, USA) and then quality control was performed (concentration, purity and integrity). Then cDNA was reversely transcribed using PrimeScript
Characteristics of AML patients and controls in RNA sequencing stage and qPCR validation stage
Characteristics of AML patients and controls in RNA sequencing stage and qPCR validation stage
AML, acute myeloid leukemia; WBC, white blood cell; FAB classification, French-American-British classification systems; NK, normal karyotype; CK, complex karyotype; FLT3-ITD, internal tandem duplications in the FMS-like tyrosine kinase 3; CEBPA, CCAAT/enhancer-binding protein
AML cell lines including AML-193, KG-1, and HL-60 were purchased from Cell Biology of the Chinese Academy of Sciences (Shanghai, China) or American Type Culture Collection (ATCC) (Manassas, USA), while normal control cells were obtained by isolating CD34
Transfection and assays
LncRNA RP11-342M1.7 small interference RNA (siRNA) and blank siRNA were synthesized and trans- fected into KG-1 cells as lncRNA RP11-342M1.7 (-) and control (-) group respectively. At 24 hours post-transfection, qPCR was used to detect lncRNA RP11-342M1.7 expression in each group. At 0, 24, 48 and 72 hours post-transfection, cell proliferation ability in each group was determined by CCK8 assay. Cell apoptosis rate in each group was measured by AV/PI assay at 48 hours post transfection.
CCK-8 assay
10 ul of CCK-8 (Dojindo, Japan) and 90 ul of medium were added to each group of KG-1 cells, and cells were incubated at 37
PCA plot analysis. LncRNAs (A) and mRNAs (B) which were detected in 50% or above samples were included in PCA analysis, which showed that lncRNAs and mRNAs expressions were able to distinguish AML patients from controls. PCA was performed using Stats packages. PCA, principal component analysis; lncRNAs, long non-coding RNAs; mRNAs, messenger RNAs.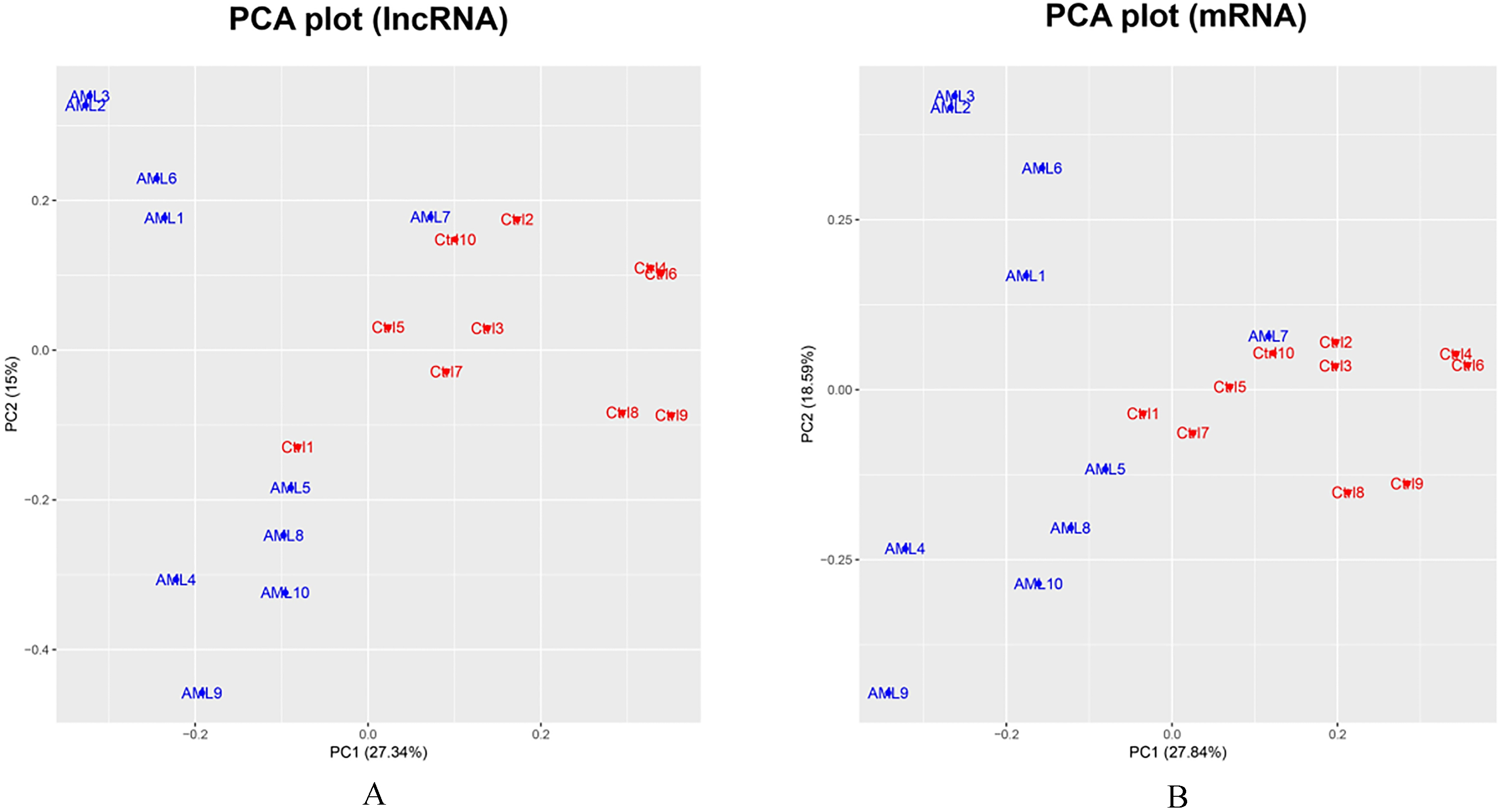
KG-1 cells were digested using pancreatin and washed with phosphate buffer solution (PBS), then suspended in 100 ul blinding buffer. Following that, 2 ul AV (Invitrogen, USA) was added to the cells and the cells were placed on ice in the dark for 15 mins. Subsequently, 1 ul PI (Invitrogen, USA) was added, and cell apoptosis rate was analyzed using flow cytometry (FCM) (Becton Dickinson, USA).
Statistics
Data were presented as mean
Results
Baseline characteristics at RNA sequencing stage and qPCR validation stage
At RNA sequencing stage, 10 AML patients with mean age of 43.9
PCA plot analysis of lncRNAs and mRNAs
LncRNA and mRNA that were detected in 50% or above samples were included in the analysis. In PCA plot analysis, lncRNA expression patterns were able to differentiate AML patients from controls to some extent (Fig. 1A); and the mRNA expression patterns could tell AML patients from controls as well (Fig. 1B). However, AML patients presented greater variation than that of controls, and sample AML7 was an outlier.
Volcano and heatmap analysis. The volcano plot showed 216 upregulated and 412 downregulated lncRNAs (A) as well as 665 upregulated mRNAs and 885 downregulated mRNAs (C) in AML compared with controls. The statistical significance was defined as 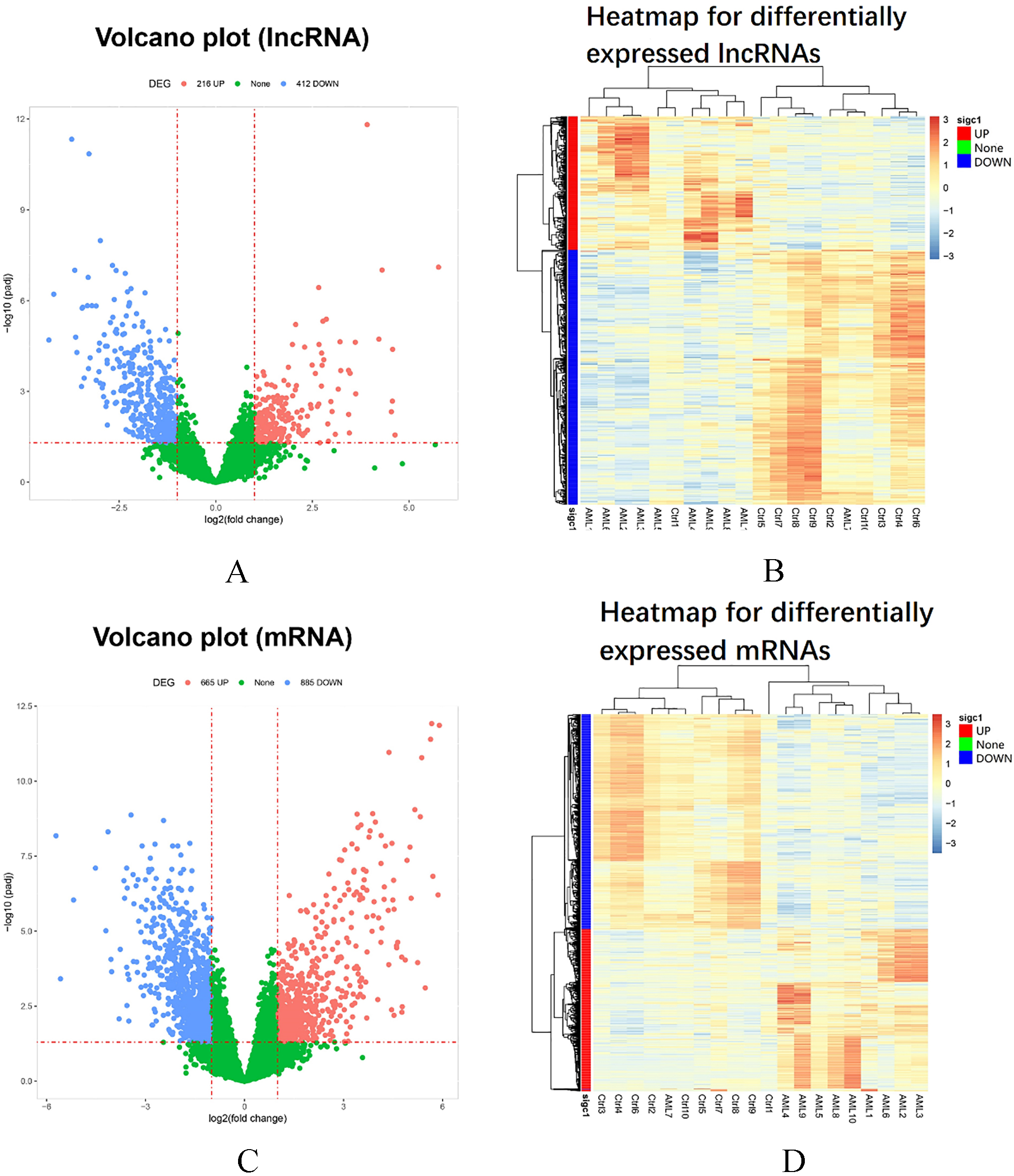
GO and KEGG enrichment analysis. The molecular functions (green bars), cellular components (blue bars) and biological processes (red bars) that differentially expressed lncRNAs were enriched in were presented by GO enrichment analysis by trans targets (A) and GO enrichment analysis by cis targets (C), and the pathways these differentially expressed lncRNAs were involved in were exhibited by KEGG enrichment analysis by trans targets (B) and KEGG enrichment analysis by cis targets (D). As for the differentially expressed mRNAs, the molecular functions (green bars), cellular components (blue bars) and biological processes (red bars) they were enriched in were shown by GO enrichment analysis (E), and the pathways they were implicated in were shown by KEGG enrichment analysis (F). GO, Gene Ontology; KEGG, Kyoko Encyclopedia of Genes and Genomes; LncRNAs, long non-coding RNAs; mRNAs, messenger RNAs.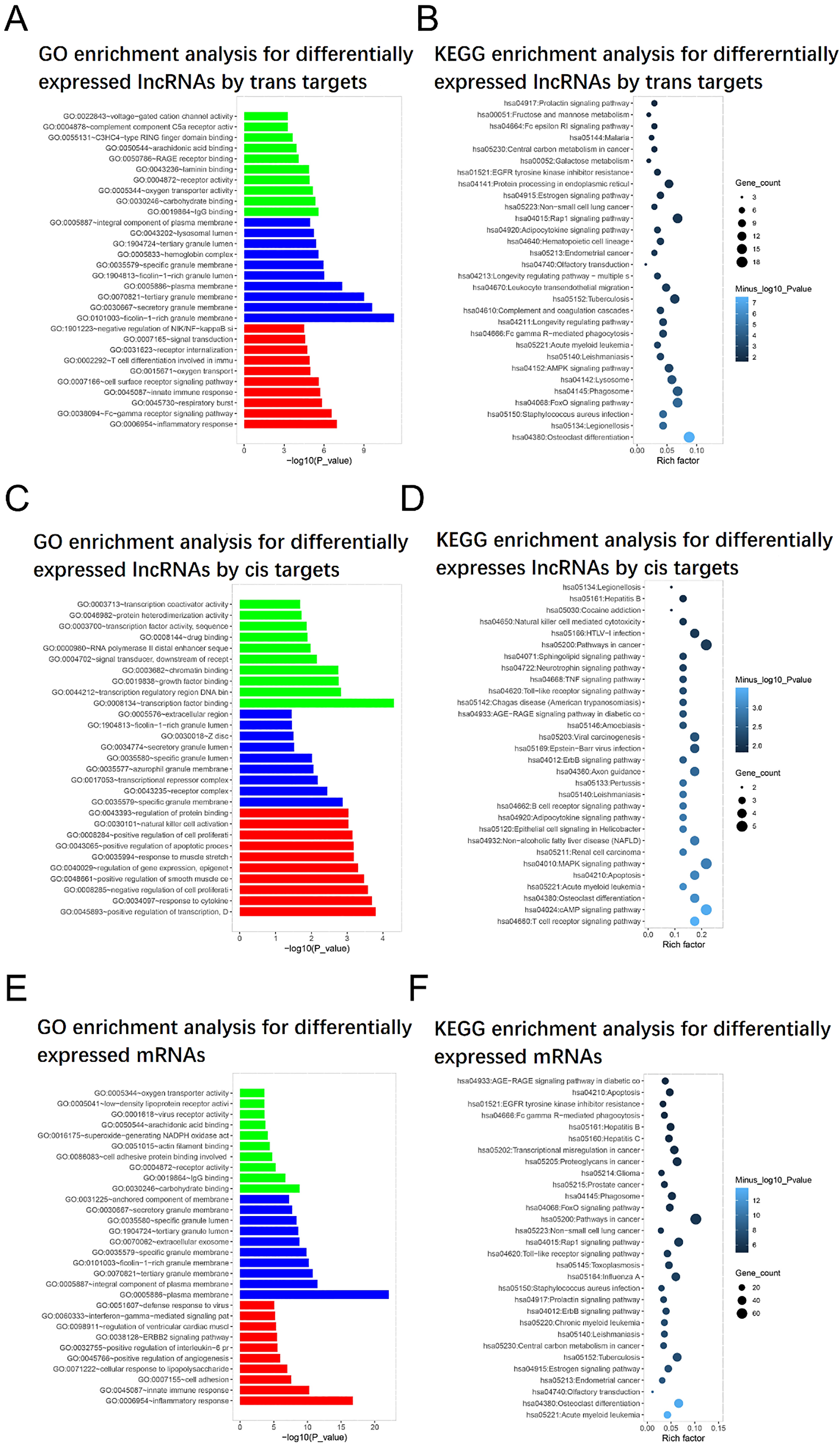
The volcano plot of lncRNAs showed that there were 216 upregulated and 412 downregulated lncRNAs in AML patients compared with controls (Fig. 2A). The threshold was set to log2 fold change and adjusted
GO and KEGG enrichment analysis of lncRNAs and mRNAs
The biological processes, cellular components and molecular functions that differentially expressed lncRNAs and mRNAs were implicated in were presented by GO enrichment analysis, and the pathways that these differentially expressed lncRNAs and mRNAs involved in were exhibited by KEGG enrichment analysis. GO enrichment analysis by trans target revealed that differentially expressed lncRNAs were enriched in molecular functions (e.g. IgG binding, carbohydrate binding and oxygen transporter activity), cellular components (e.g. ficolin-1-rich granule membrane, secretory granule membrane and plasma membrane) and biological processes (e.g. inflammatory response, Fc-gamma receptor signaling pathway and innate immune response) (Fig. 3A). KEGG enrichment analysis by trans target displayed that differentially expressed lncRNAs participated in neoplastic pathways such as FoxO signaling pathway, AMPK signaling pathway and Rap1 signaling pathway etc. (Fig. 3B). And in GO enrichment analysis by cis target, the differentially expressed lncRNAs were enriched in molecular functions (e.g. transcription factor binding, transcription regulatory region DNA binding and growth factor binding), cellular components (e.g. specific granule membrane, receptor complex and transcriptional repressor complex) and biological processes (e.g. response to cytokine, negative regulation of cell proliferation and positive regulation of apoptotic process) (Fig. 3C). From KEGG enrichment analysis by cis target, the differentially expressed lncRNAs participated in leukemia-associated signaling pathways such as cAMP signaling pathway, T cell receptor signaling pathway and MAPK signaling pathway etc. (Fig. 3D). As for differentially expressed mRNAs, GO enrichment analysis disclosed that they were enriched in molecular functions (e.g. carbohydrate binding, IgG binding and receptor activity), cellular components (e.g. plasma membrane, integral component of plasma membrane and tertiary granule membrane) and biological processes (e.g. inflammatory response, innate immune response and cell adhesion) (Fig. 3E), and KEGG enrichment analysis showed that the differentially expressed mRNAs were associated with pathways in AML, pathways in other cancers and osteoclast differentiation (Fig. 3F).
Regulatory network of differentially expressed lncRNAs
Since the number of trans target mRNAs for differentially expressed lncRNAs was large, only 10 upregulated and 10 downregulated lncRNAs with trans target mRNAs were presented in regulatory network (Fig. 4A). The detailed information about the top 10 upregulated and 10 downregulated lncRNAs were listed in Table 2. And due to limited cis target mRNAs of differentially expressed lncRNAs, the regulatory network of all differentially expressed lncRNAs with cis target mRNAs was shown (Fig. 4B).
Top 10 upregulated and 10 downregulated lncRNAs in AML patients compared to controls
Top 10 upregulated and 10 downregulated lncRNAs in AML patients compared to controls
Top 10 upregulated and 10 downregulated lncRNAs were selected based on the rank of absolute value of Log
Regulatory network of differentially expressed lncRNAs and their target mRNAs. The regulatory network of the top 10 upregulated and 10 downregulated lncRNAs with their trans target mRNAs (A); the regulatory network of all differentially expressed lncRNAs with cis target mRNAs were shown (B). Data were analyzed using Igraph package. Red dots stood for upregulated lncRNAs, green dots stood for downregulated lncRNAs, red squares stood for target mRNAs for upregulated lncRNAs, greed squares stood for target mRNAs for downregulated lncRNAs. LncRNAs, long non-coding RNAs; mRNAs, messenger RNAs.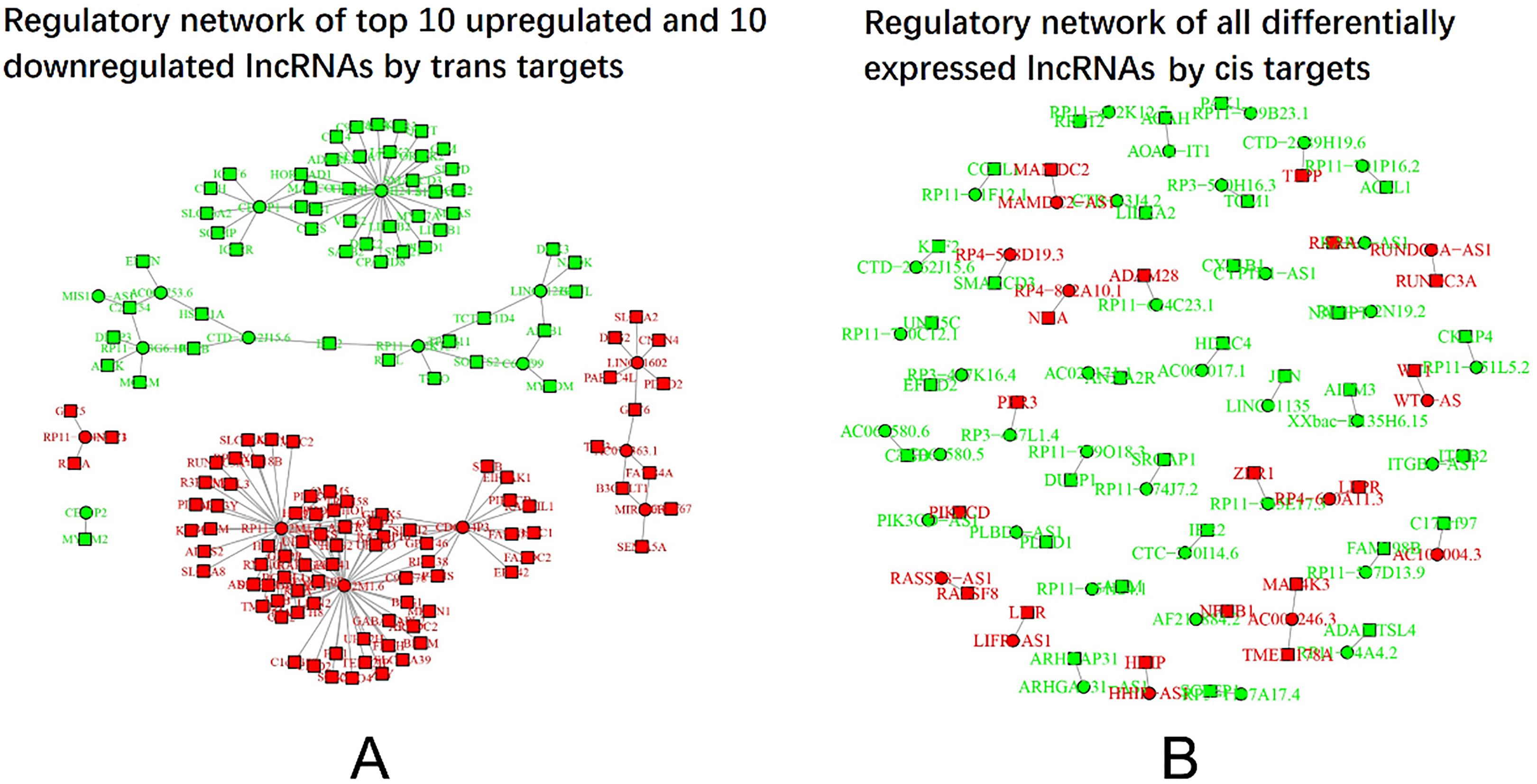
In order to comprehensively illustrate the transcription and regulation of the differentially expressed lncRNAs and mRNAs, circus graph was drawn (Fig. 5). The outermost layer represented chromosome number and the second outermost layer represented upregulated (in red color) and downregulated (in green color) mRNAs, the third outermost layer represented upregulated (in red color) and downregulated (in green color) lncRNAs. As for the collected lines in the center, they were illustrations of trans or cis regulation between lncRNAs and mRNAs.
Circos graph. Circos graph for transcription and regulation information was drawn using RCircos package. The outermost layer was the chromosomes, the second outermost layer represented lncRNAs, the third outermost layer represented mRNAs, the collected lines in the middle represented cis or trans interaction between lncRNAs and mRNAs. Green stood for downregulated genes and red stood for upregulated genes. LncRNAs, long non-coding RNAs; mRNAs, messenger RNAs.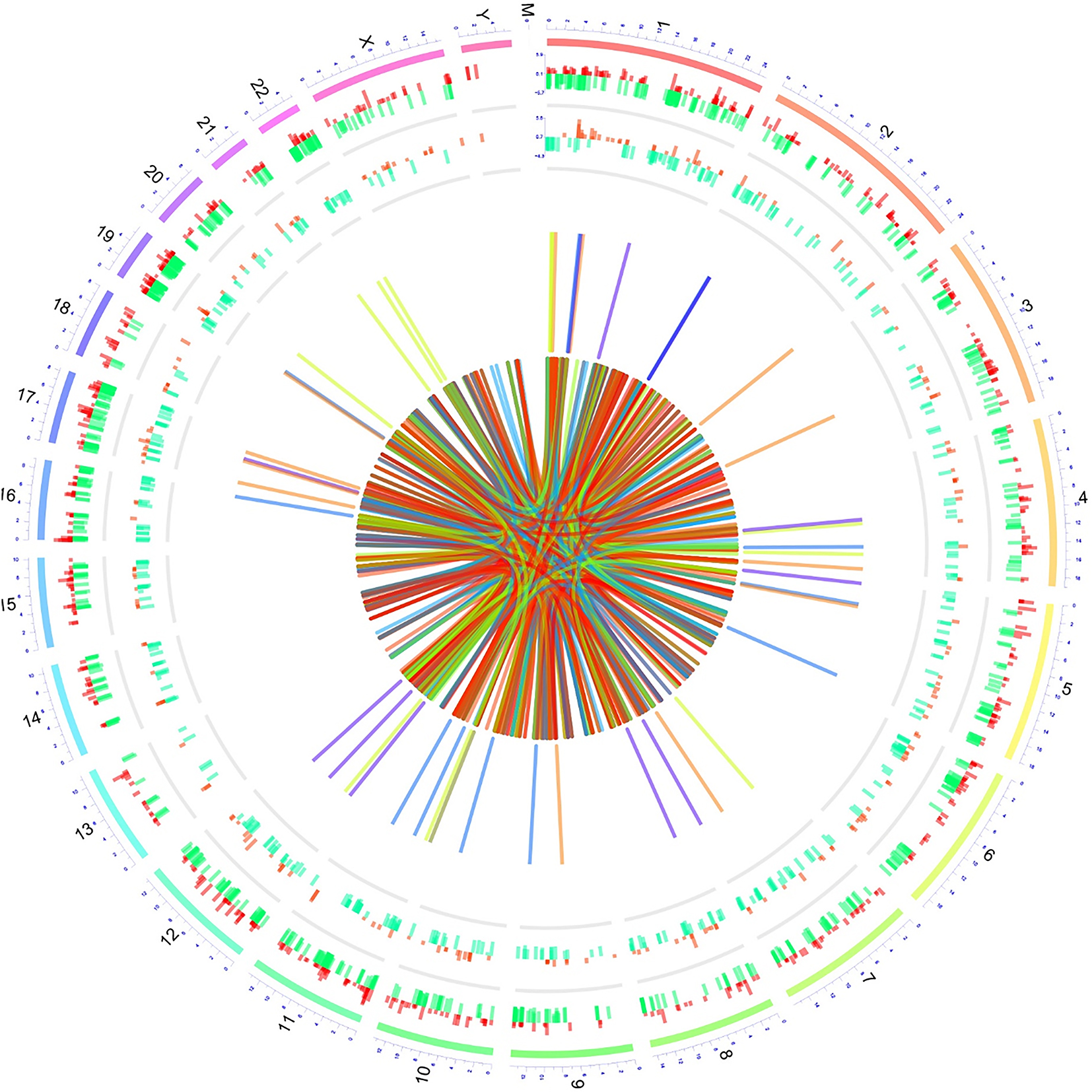
The top 5 upregulated and top 5 downregulated lncRNAs in AML patients compared to controls in RNA sequencing stage were further included in the qPCR validation stage. The expressions of these top 5 upregulated and top 5 downregulated lncRNAs were compared between AML patients and controls. Among the top 5 upregulated lncRNAs, lncRNA RP11-342M1.7 (
Logistic regression analyses of candidate lncRNAs for predicting AML risk
Logistic regression analyses of candidate lncRNAs for predicting AML risk
lncRNAs, long non-coding RNAs; AML, acute myeloid leukemia; OR: odds ratio; CI: confidence interval.
Expression of candidate lncRNAs in AML compared with controls. LncRNA RP11-342M1.7 (A) and lncRNA CDCA4P3 (C) expressions were increased in AML patients compared with controls, while lncRNA AC074363.1 (B), lncRNA LINC01602 (D) and lncRNA RP11-754N21.1 (E) expressions were similar between AML patients and controls. LncRNA CES1P1 (F), lncRNA AC008753.6 (G) and lncRNA RP11-573G6.10 (J) expressions were decreased in AML patients compared with controls, while lncRNA LINC01220 (H) and lncRNA CTD-2562J15.6 (I) expressions were similar between AML patients and controls. Comparison of candidate lncRNAs expressions between AML patients and controls was determined by Wilcoxon rank sum test. 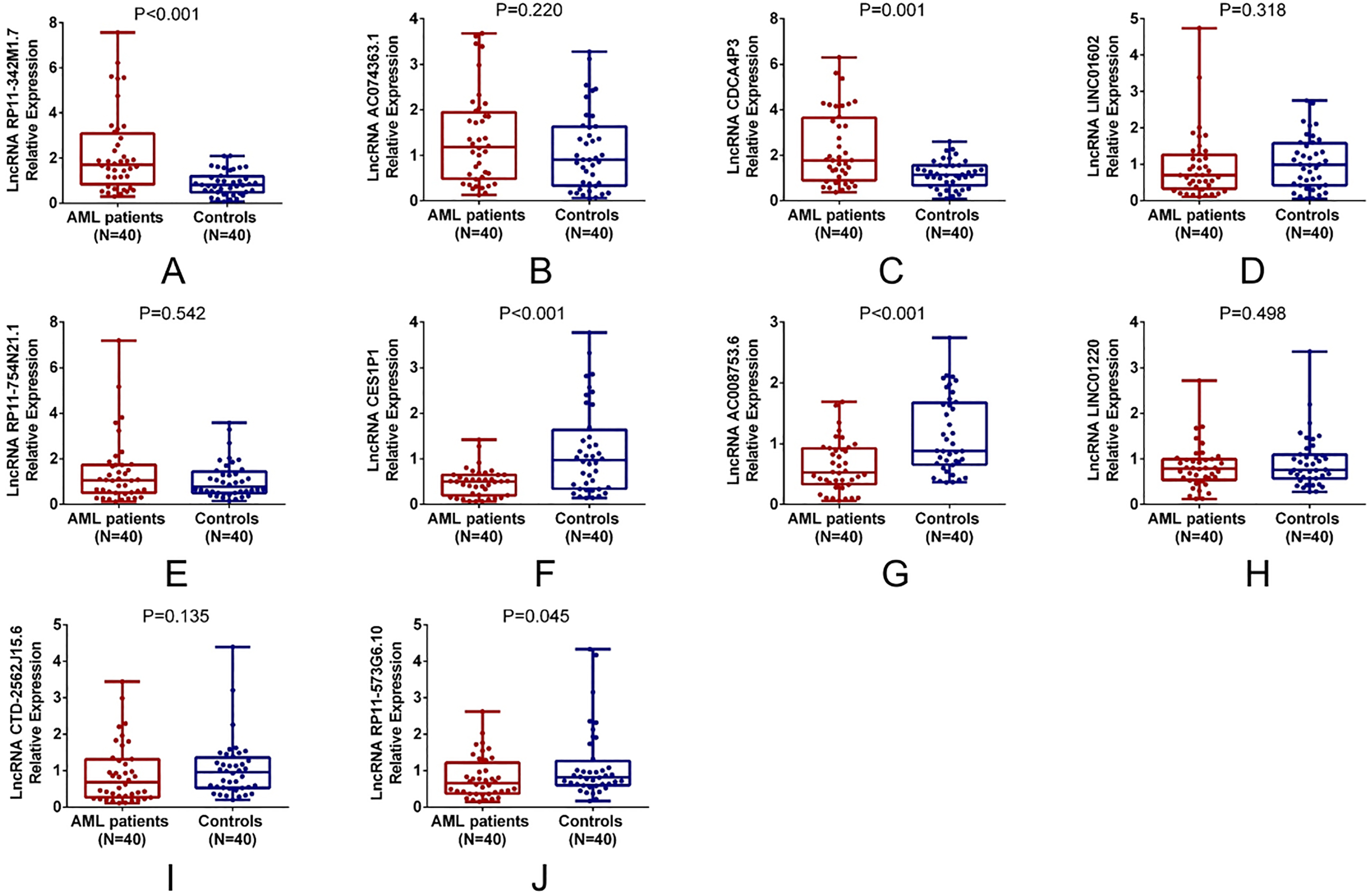
Univariate logistic regression analysis exhibited that lncRNA RP11-342M1.7 (
ROC curve of candidate lncRNAs
Candidate lncRNAs that were independent predictive factors for AML risk were further selected for ROC curve analysis, which observed that lncRNA RP11-342M1.7 (AUC: 0.768; 95% CI: 0.663–0.872), lncRNA CES1P1 (AUC: 0.729; 95% CI: 0.616–0.843) and lncRNA AC008753.6 (AUC: 0.736; 95% CI: 0.629–0.843) were of good value in distinguishing AML patients from controls (Fig. 7). And it was more important to note that the combination of these three lncRNAs was of remarkably great value in distinguishing AML patients from controls (AUC: 0.901; 95% CI: 0.835–0.966).
Predictive value of candidate lncRNAs for AML risk. lncRNA RP11-342M1.7 (AUC: 0.768; 95% CI: 0.663–0.872), lncRNA CES1P1 (AUC: 0.729; 95% CI: 0.616–0.843) and lncRNA AC008753.6 (AUC: 0.736; 95% CI: 0.629–0.843) were of good predictive value for AML risk. And the combination of these three lncRNAs was of remarkably great value in distinguishing AML patients from controls (AUC: 0.901; 95% CI: 0.835–0.966). Independent lncRNAs for predicting AML risk were included in ROC curves and feasibility was determined by the AUC. ROC, Receiver-operating characteristic; AUC, area under curve; lncRNAs, long non-coding RNAs; AML, acute myeloid leukemia.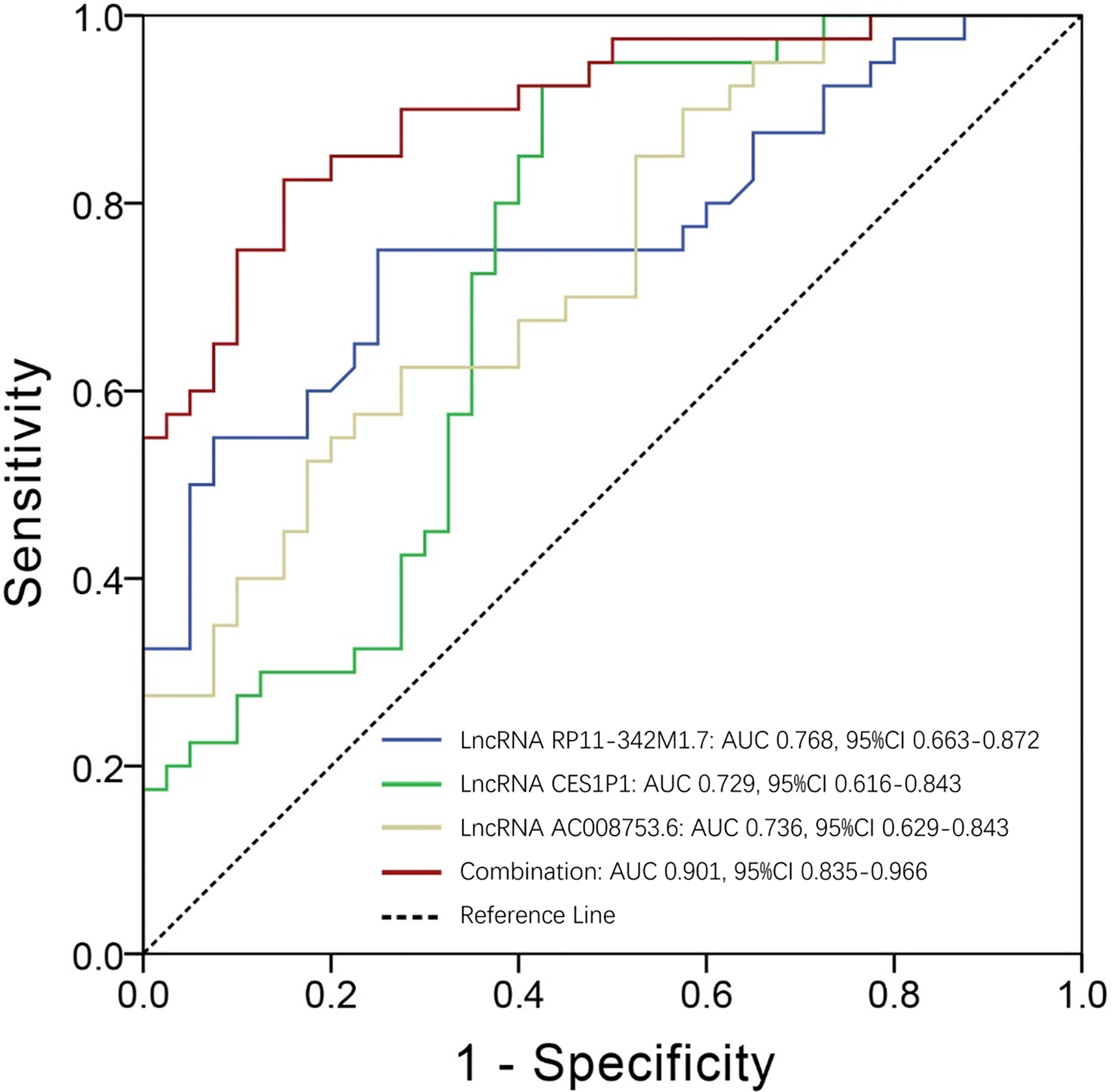
There were 9 patients did not achieve CR, and 31 patients achieved CR. LncRNA RP11-342M1.7 relative expression was increased in non-CR patients compared with CR patients (
Comparison of candidate lncRNA expressions between CR and non-CR patients. LncRNA RP11-342M1.7 (A) was increased, lncRNA AC008753.6 (G) and lncRNA CTD-2562J15.6 (I) was decreased in non-CR patients compared with CR patients. LncRNA AC074363.1 (B), lncRNA CDCA4P3 (C), lncRNA LINC01602 (D), lncRNA RP11-754N21.1 (E), lncRNA CES1P1 (F), lncRNA LINC01220 (H) and lncRNA RP11-573G6.10 (J) relative expressions were similar between non-CR and CR patients. Comparison of candidate lncRNAs expressions between CR patients and non-CR patients was determined by Wilcoxon rank sum test. 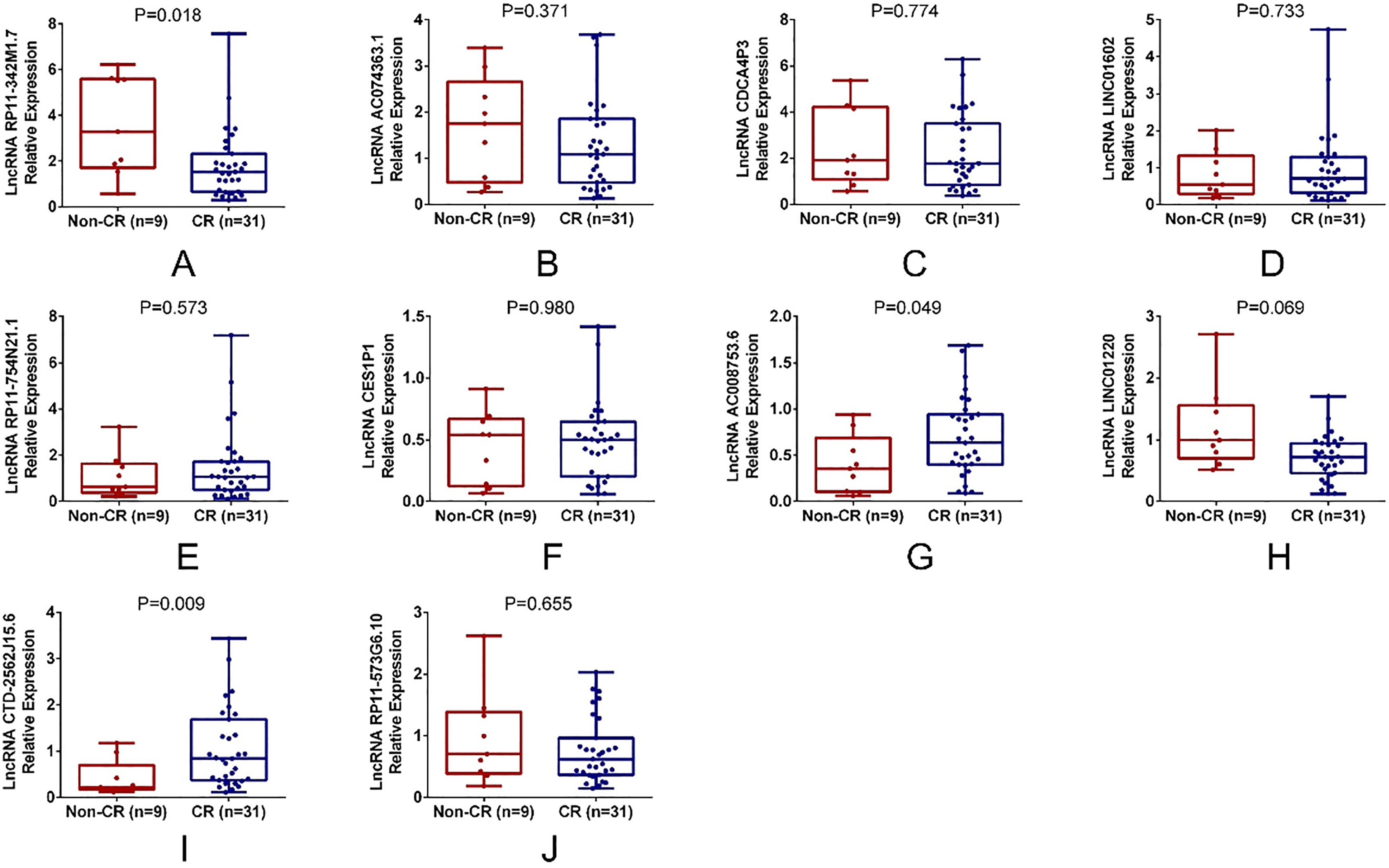
LncRNA RP11-342M1.7 relative expression was increased in AML cell lines including AML-193 (
Effect of lncRNA RP11-342M1.7 knockdown on AML cell activities. LncRNA RP11-342M1.7 was increased in AML-193, KG-1 and HL-60 cells compared with control cells (A). LncRNA RP11-342M1.7 expression was lower in lncRNA RP11-342M1.7 (-) group compared with control (-) group (B). Cell proliferation was suppressed in bu lncRNA RP11-342M1.7 knockdown at 48 hours and 72 hours after transfection (C). And cell apoptosis was promoted by lncRNA RP11-342M1.7 knockdown at 72 hours post transfection (D, E). Comparison was conducted using 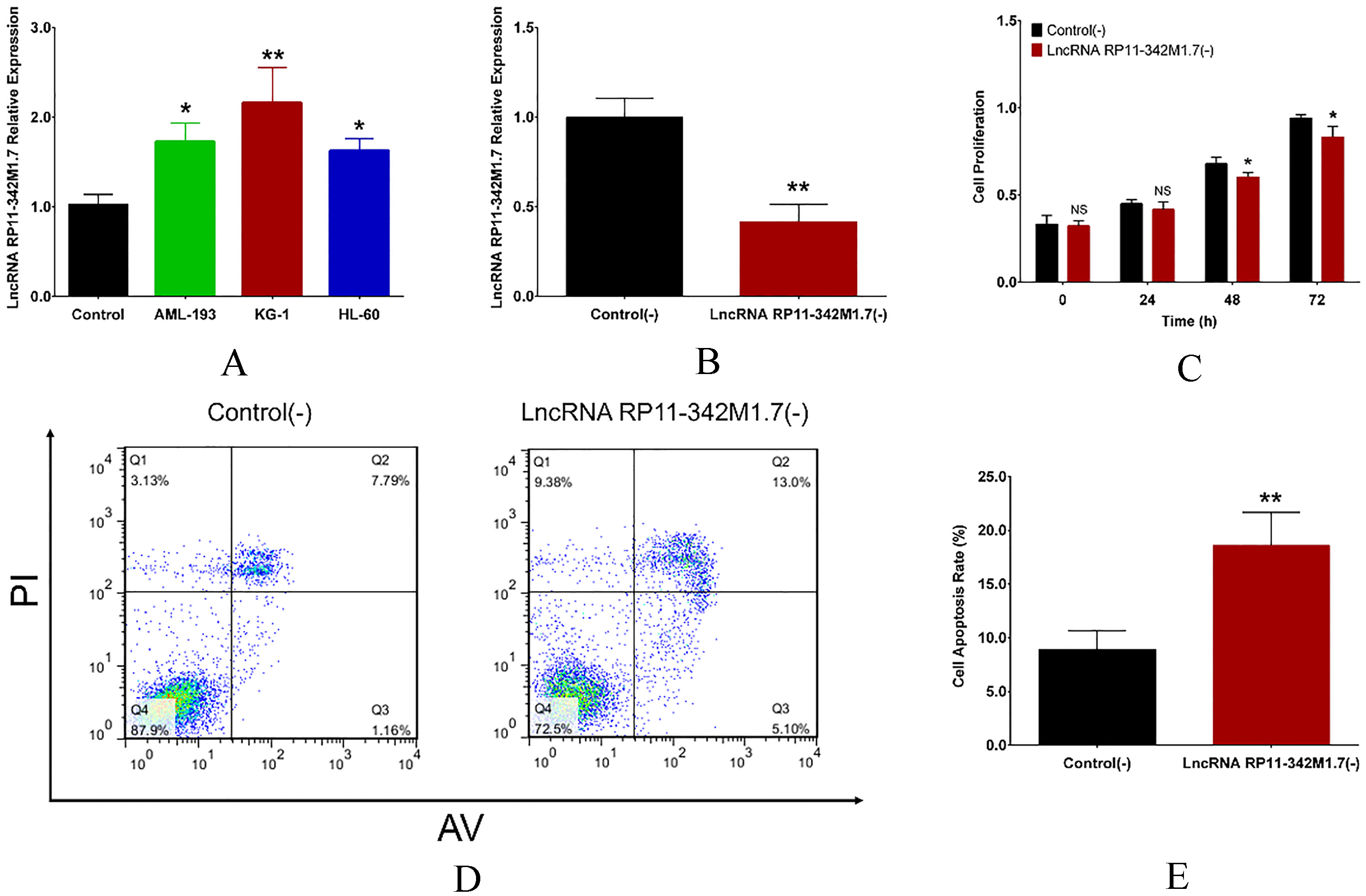
The abundant number, wide-ranged expression patterns, tumor specificity and stability of lncRNAs have made them a novel research field in cancer. And benefiting from the development of next-generation RNA sequencing, comprehensive profiling of lncRNA expression in cancers as well as the extensive bioinformatics analyses are made possible [10, 11, 12]. Relevant researches have disclosed that the aberrant expressions of lncRNAs are shown to play important roles in the occurrence and development of some solid tumors such as cervical cancer and gallbladder carcinoma [13, 14]. As for hematological malignancies, a total of 1053 lncRNAs were dysregulated in DLBCL cells lines (OCI-LY19, SU-DHL-1, OCI-LY10 and U2932) compared with normal B cells [15]. Additionally, 2560 novel lncRNAs are identified to be differentiative for T cell acute lymphoblastic leukemia cell lines (DND41, Jurkat and RPMI-8402) [6]. These evidences reveal the aberrant expression of lncRNAs in hematological malignancies, however, there is limited study that extensively investigates the lncRNAs expression profiles in AML. Although one previous study has used custom microarray platform to reveal the lncRNA expression patterns in AML, only the correlation of certain individual lncRNAs with clinical features and recurrent mutations of cytogenetically normal AML is assessed, while the detailed bioinformatic analyses about the differentially expressed lncRNAs are not included [16]. The main goal of this study was to achieve a deeper understanding of the expression profiles of lncRNAs in AML and explore the roles of differentially expressed lncRNAs, therefore, this current work assessed the lncRNA expression profiles, and conducted bioinformatic analyses that disclosed the functions of the aberrantly expressed lncRNAs. We observed 216 upregulated and 412 downregulated lncRNAs as well as 665 upregulated and 885 downregulated mRNAs in AML patients compared with controls. In addition, these lncRNAs and mRNAs were shown to be involved in neoplastic signaling pathways such as cAMP signaling pathway, MAPK signaling pathway and AMPK signaling pathway, which indicated the functions of differentially expressed lncRNAs in pathogenesis of AML. It was also interesting to mention that due to the heterogeneity of AML as a disease, the variation among AML samples is greater than that within the control group. And the outlier AML7 in PCA analysis might be attributed to the heterogeneity of AML or because of less apparent disease properties in this particular patient.
Currently, a variety of specific lncRNAs are known to be implicated in hematological malignancies, and a few of them are associated with disease risk and might potentially serve as diagnostic biomarkers or therapeutic targets in AML [4]. For example, lnc-SOX6-1 expression is upregulated in pediatric AML patients compared with healthy controls, and associates with monosomal karyotype as well as severe risk stratification [17]. Another frequently studied lncRNA TUG1 is shown to be upregulated in AML compared with healthy controls and predict increased WBC count as well as higher risk stratification [3]. The existing evidence indicated the levels of several individual lncRNAs in AML and their correlation with risk stratification as well as clinicopathological features. However, there are still a huge number of lncRNAs that remain unannotated in AML, and their predictive value for AML risk are still obscure. In qPCR validation stage of our study, the top 5 upregulated and top 5 downregulated lncRNAs in AML compared with controls were selected from the RNA sequencing stage, and their predictive value for AML risk were further evaluated in 40 AML patients and 40 age and gender matched controls. We observed that lncRNA RP11-342M1.7 and lncRNA CDCA4P3 were upregulated, while lncRNA CES1P1, lncRNA AC008753.6 and lncRNA RP11-573G6.10 were downregulated in AML patients compared with controls. In addition, lncRNA RP11-342M1.7, lncRNA CES1P1 and lncRNA AC008753.6 were independent predictive factors for AML susceptibility, and most importantly, the combination of these three lncRNAs was of remarkably good value in distinguishing AML patients from controls. This could be explained by: (1) lncRNA RP11-342M1.7, lncRNA CES1P1 and lncRNA AC008753.6 might be implicated in AML pathogenesis via regulating degradation and translation of their target mRNAs (as shown in regulatory network Fig. 4), thereby altered disease risk of AML [18]. For instance, lncRNA RP11-342M1.7 might target mRNA GMPR (ENSG00000137198) (derived from RNA sequencing analysis) that was implicated in differentiation of leukemia cells [19, 20]. (2) They could also recruit transcription factors or epigenetic modification complexes which regulated transcriptional activities of parental genes in cis or trans manner, and influenced various cellular functions such as immune responses and inflammatory responses, thereby affecting AML susceptibility. However, the detailed mechanisms of these lncRNAs in the pathogenesis of AML should be further explored. Therefore, we performed cellular experiments and disclosed that lncRNA RP11-342M1.7 knockdown suppressed cell proliferation but promoted cell apoptosis in AML cells.
In the present study, we evaluated the lncRNA expression patterns in AML for the first time and revealed the predictive value of several candidate lncRNAs for AML risk, whereas there were still some weaknesses to this work. Since this was an explorational study, we only pointed out the possibility of several lncRNAs as biomarkers for AML, however, it would certainly need a greater effort in the future to validate the diagnostic and prognostic value of these lncRNAs and solve real clinical problems. Besides, the number of total participants was 40 AML patients plus 40 age and gender matched controls in qPCR validation stage, which was relatively small, however, in RNA sequencing stage, 10 AML patients and 10 age and gender matched controls were included, which was a greater sample size compared with that in most of the previous studies. Additionally, the detailed mechanism of candidate lncRNAs and their target mRNAs in AML was not investigated.
In conclusion, our study reveals the comprehensive lncRNAs expression profiles in AML, and proposes candidate lncRNAs including lncRNA RP11-342M1.7, lncRNA CES1P1 and lncRNA AC008753.6 that are with independent predictive potential for AML risk, which might serve as reference for further research about lncRNAs in AML.
Footnotes
Conflict of interest
The authors declare no conflict of interests.
Supplementary
Primers list
Gene
Forward primer (5’-3’)
Reverse primer (5’-3’)
LncRNA RP11-342M1.7
ACTCCTTCCACTTGCTCATCCA
GCTCCGCTTCTCCTCCTCTT
LncRNA AC074363.1
AGCGGAGCAGCAGCATCTT
AGGTTGTCACACACTGTCTTCA
LncRNA CDCA4P3
GCCTCAGATGTGGTCAGTTCTT
CTCTTGCTTGCCCAGGTCAG
LncRNA LINC01602
CACAGTTAGTTGCTTCACATTGAC
TCTTCTCAGGTATAACAGACTCAGT
LncRNA RP11-754N21.1
ACTTGCTGTAGAAGGTGTGAAGG
TCTGCTGTCTGGATGATGGTTAC
LncRNA CES1P1
TAGAATCACTGAGGCACCAATG
AGAGACACAAGACACCATCACT
LncRNA AC008753.6
GGAGAAGGAAATCGGGACTGG
GCTCCTTGCCTGTAAGATTGC
LncRNA LINC01220
AGGCAACTCGGACTAAGAAGAC
GGCTGCTGTAACTCCAAGAAC
LncRNA CTD-2562J15.6
CTTGGCTGTGCTTCCTGCTA
ACTTCTCTGTTCCTCTGTGGTG
LncRNA RP11-573G6.10
GGCTTCAGGCTCTTTCTTGCT
CTGTATCAGCTATTGGCTCCTATCC
GAPDH
GACCACAGTCCATGCCATCAC
ACGCCTGCTTCACCACCTT