
Abstract
BACKGROUND:
Worldwide, cervical cancer is the fouth leading cause of deaths in gynecological oncology. Although the causes of cervical cancer have been extensively investigated, understanding of its exact pathogenesis remains incomplete.
OBJECTIVE:
This study aimed to identify alterations of genome and transcriptome of HPV associated cervical cancer pathogenesis using multi-omics approaches.
METHODS:
Cervical cancer and matched adjacent non-tumor specimens of one HPV16+ and two HPV- patients were sampled for whole-exome sequencing (WES) and RNA sequencing to characterize DNA mutations and gene expression profiles. WES and Affymetrix SNP 6.0 arrays data were analyzed from 6 HPV- and 93 HPV16+ cervical cancer patients in the cancer genome atlas (TCGA) database, as an independent validation group.
RESULTS:
WES identified 64 somatic mutation genes in tumors of 3 patients. HPV16+ tumor got fewer somatic mutated genes than HPV- tumors, which was validated by TCGA results. In this study, somatic mutated profile, CNV and gene expression heat map presented that HPV16+ tumors was distinct with HPV- tumors. The most significant altered pathways and GO terms were both related with cell cycle. Integrated analysis of multi-omics showed positive correlation between gene expression level and copy numbers.
CONCLUSIONS:
The results of this study provided novel insights into the pathogenesis of HPV associated cervical cancer.
Introduction
Cervical cancer is the fouth most common malignancy and the fouth leading cause of deaths in gynecological oncology worldwide [1]. Cervical cancer predominantly occurs between the ages of 40–55 years; however, the disease is still detected in women over 80-years-old [2]. More than half a million women are diagnosed with cervical cancer every year worldwide, and mortality rates are especially high in less-developed countries [3]. Cervical cancer screening based on human papillomavirus (HPV) testing has proven effective for reducing the incidence of cervical cancer [4]. The current clinical management for cervical cancer mainly depends on surgery, chemotherapy, and radiotherapy. The limited number of clinical trials testing the efficacy of targeted therapy against cervical cancer, although encouraging, have shown limited effects on survival [5, 6]. Although cervical cancer pathogenesis has been extensively investigated, the exact mechanisms of carcinogenesis remain unclear [7].
Genetic mutations have been found to play critical roles in the risk of developing cervical cancer [8]. A recent sequencing study based on exomes, genomes, and transcriptomes identified 13 recurrently mutated genes [9]. Another comprehensive analysis of cervical cancer, The Cancer Genome Atlas (TCGA), revealed new genetic mutations that might serve as biomarkers for identifying clinically important subgroups of cervical cancer patients [10]. Additionally, previous studies have demonstrated the important roles of long non-coding RNAs, circular RNAs, microRNAs and mRNAs in cervical cancer development and progression [11, 12, 13].
High-throughput sequencing provides a valuable platform for cervical cancer research; however, most previous studies were performed using a single omics technique. There has been a lack of studies using a combined omics approach to elucidate cervical cancer pathogenesis. In this study, a combination of whole-exome and RNA sequencing was employed to identify the key genes and biological pathways involved in cervical cancer development to provide new insights into cervical cancer pathogenesis.
Materials and methods
Patients and samples
Tumors and paired adjacent non-tumor specimens were sampled from three cervical carcinoma patients during surgery or biopsies in Quanzhou First Hospital. All histopathologic diagnoses of cervical carcinoma were confirmed by three pathologists. All specimens were snap-frozen in liquid nitrogen immediately after resection. Demographical and clinical data were collected from each patient.
This study was approved by the Ethics Review Committee of Quanzhou First Hospital Affiliated to Fujian Medical University (permission no. FZDYYY2013-00121). Written informed consent was obtained from all subjects participating in the study, following a detailed description of the purpose of the study. The study conforms with The Code of Ethics of the World Medical Association (Declaration of Helsinki), printed in the British Medical Journal (18 July 1964).
Sequence data generation
Whole exome-sequencing (WES)
Genomic DNA was extracted from tissues using the QIAamp DNA Mini Kit (Qiagen; Hilden, Germany). DNA enrichment was performed in the whole exon areas with an Agilent liquid chip capture system (Agilent Technologies; Santa Clara, CA, USA), followed by high-throughput sequencing on an Illumina platform (Illumina, Inc.; San Diego, CA, USA). Briefly, 1
RNA sequencing
Total RNA was extracted from the tissues, and RNA degradation and contamination was monitored on 1% agarose gels. RNA purity was checked using the NanoPhotometer
Approximately 1.5
Clustering of the index-coded samples was performed on a cBot Cluster Generation System using HiSeq 2500 PE Cluster Kit (Illumina) according to the manufacturer’s instructions. After cluster generation, the library preparation was sequenced on an Illumina Hiseq 2500 platform (Illumina), and 125 bp paired end reads were generated.
Bioinformatics analysis
Quality control
Raw sequencing reads were filtered according to the following criteria: (1) reads containing sequencing adaptors were removed; (2) reads containing more than 10% N; and (3) low quality bases (
Clinical characteristic of cervical patients enrolled in this study
Clinical characteristic of cervical patients enrolled in this study
SCC, squamous cell carcinoma; AUC, adenocarcinoma of the uterine cervix; FIGO, International Federation of Gynecology and Obstetrics. HPV, Human Papilloma Virus.
Reads were aligned to the reference human genome (UCSC hg19;
Somatic SNVs and InDels of tumors compared with matched normal tissue were named and functionally annotated using MuTect v. 1.1.4 and Varscan2 v. 2.3.9 software. Variants were annotated using the ANNOVAR software tool. Annotations for mutation function, mutation location, amino acid changes, 1000 Genomes Project data and dbSNP reference number were performed. Somatic mutations were filtered by criterion of frequency
Quality statistics of whole-exome sequecing and RNA sequencing data
Quality statistics of whole-exome sequecing and RNA sequencing data
Differential expression analyses of cervical cancer and adjacent non-tumor tissues were performed using the Cuffdiff program. The selection criteria included a false discovery rate (FDR) of
DEGs were submitted to IPA (Ingenuity Pathway Analysis), and functional annotation identified the biological functions that were most significant in the dataset. A Fisher’s exact test was used to calculate a
Data of cervical cancer in TCGA cohort
Whole-exome sequencing and Affymetrix SNP 6.0 arrays data of 93 HPV16+ and 6 HPV-cervical cancer patients of TCGA study were downloaded in Broad GDAC Firehose (http://gdac.broadinstitute.org). Then somatic mutations and copy number variations were analyzed and compared between HPV- and HPV16+ cervical cancer.
Results
Patients and sequencing data
Table 1 showed the clinical characteristics of three cervical cancer patients enrolled in this study. Histological type of two patients was squamous cell carcinoma (SCC), and one was adenocarcinoma of the uterine cervix (AUC). One patients was infected with human papilloma virus (HPV) 16 type. FIGO stage of tumors were IIB, IB2 and IIB, respectively.
Somatic mutations in cervical cancer. (A) Somatic mutation types in tumors of three cervical cancer patients; (B) Venn diagram of somatically mutated genes among tumors of three cervical cancer patients. (C) Somatic mutation counts of HPV- and HPV16+ cervical cancer in TCGA cohort. (D) Venn diagram of somatic mutated genes between HPV- and HPV16+ cervical cancer in TCGA cohort. (E) Enrichment of specific mutated genes of HPV- and HPV16+ tumors in TCGA cohort by IPA.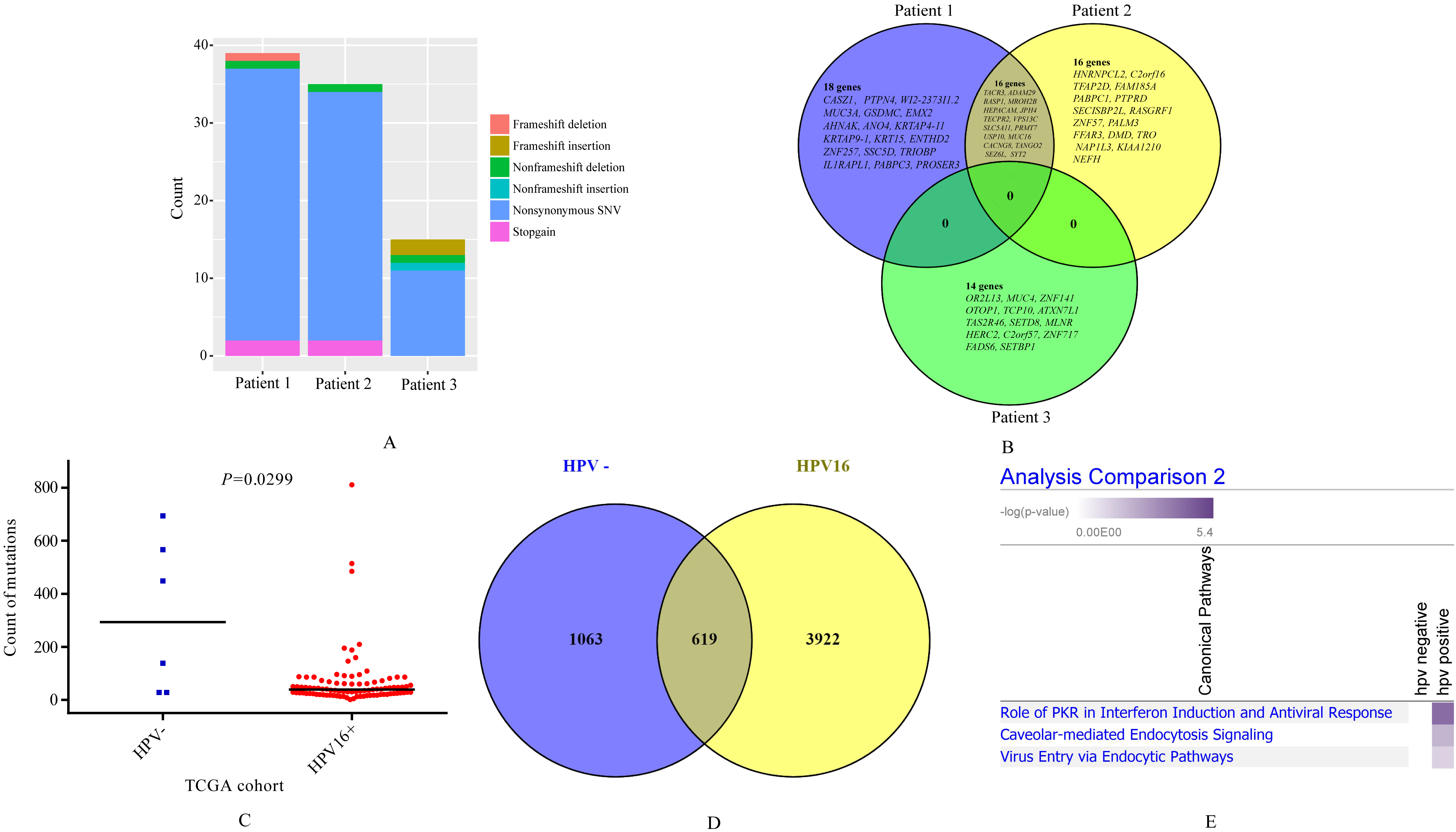
Table 2 showed quality statistics of sequencing data. WES generated mean 6.11 GB clean data, with 0.93 of Q30, and mean 65.21 coverage of sequencing depth of tumors and normal tissues. The mean GC percentage was 49.92. RNA sequencing produced average of 6.85 GB clean data, with 95.37% of Q30, and 86.48% mapped rate with reference genome.
Sixty-four somatic mutated genes were identified from three cervical cancer patients (Supplementary Table 1). We found mutation count of patient 3 was far less than patient 1-2, and mutation type was also different (Fig. 1A). Venn diagram compared mutated genes among three patients (Fig. 1B). We found 16 overlapped mutated genes between patient 1 and patients 2, including SYT2, TACR3, ADAM29, BASP1, MROH2B, HEPACAM,
USP10, et al. But no overlaps of somatic mutated genes was found between HPV16+ patient and two HPV-patients (Fig. 1B). Similar feature was found in somatic mutation genes in TCGA cohort. HPV negative tumors harbored more somatic mutations than HPV16+ tumors (
Then we compared somatic mutation genes in this study with TCGA result. Majority of 64 somatic mutated genes were reported by TCGA study (Supplementary Table 1). We found 13 novel somatic mutated genes which were not reported by TCGA study, including SYT2, BASP1, WI2-2373I1.2, JPH4, KRTAP9-1, HNRNPCL2, OR2L13, TAS2R46, and so on (Supplementary Table 1). However, the recurrent somatic mutated genes in cervical cancer such as PIK3CA, TTN, and FBXW7 were not identified in this study, which may result from small sample size for WES in this study.
Somatic mutation genes of HPV associated cervical cancer
Interestingly, patient 3 was infected with HPV16, whose tumor presented entirely different mutated profiling compared with other two HPV negative patients. All 14 mutated genes in tumors of patient 3 were not found in other two patients, including OR2L13, MUC4, ZNF141, OTOP1, TCP10, et al (Fig. 1B). TCGA study reported mutated frequency of MUC4 was 17.2%, which was the top 4 frequency mutated gene of cervical cancer (cBioportal,
We performed enrichment of specific mutated genes of HPV- and HPV16+ tumors in TCGA cohort by IPA. From Fig. 1E, we can see somatic mutated genes in HPV16+ tumors specifically enriched to virus-infection related pathways including role of PKR in interferon induction and antiviral response, caveolar-mediated endocytosis signaling, and virus entry via endocytic pathways, compared with HPV-tumors.
CNV analysis
Copy number variations (CNVs) were plotted by chromosomal location using CNVkit and GISTIC2.0 software of the WES dataset of three patients (Fig. 2A) and TCGA cohort (Fig. 2B), respectively. Tumors of patient 1 and patient 2 showed similar CNV heat map, but distant from patient 3. We found focal amplification of 1q21-31, 1q41-44, and 3q22-29 in tumors of patient 1 and patient 2, and scattered amplifications and deletions in other chromosomes. In TCGA cohort, HPV16+ tumors exhibited similar whole CNV profile, but different parts (Fig. 2B). Focal amplifications and deletions were showed in Supplementary Fig. 1 of TCGA result. HPV16+ tumors harbored less CNVs than HPV- tumors in this study, while, which was not validated by TCGA result.
Copy number variations (CNVs) of tumors are plotted by chromosomal location (vertical axis) of the entire dataset. (A) Tumors of 3 patients in this study. (B) Cervical cancer of TCGA cohort.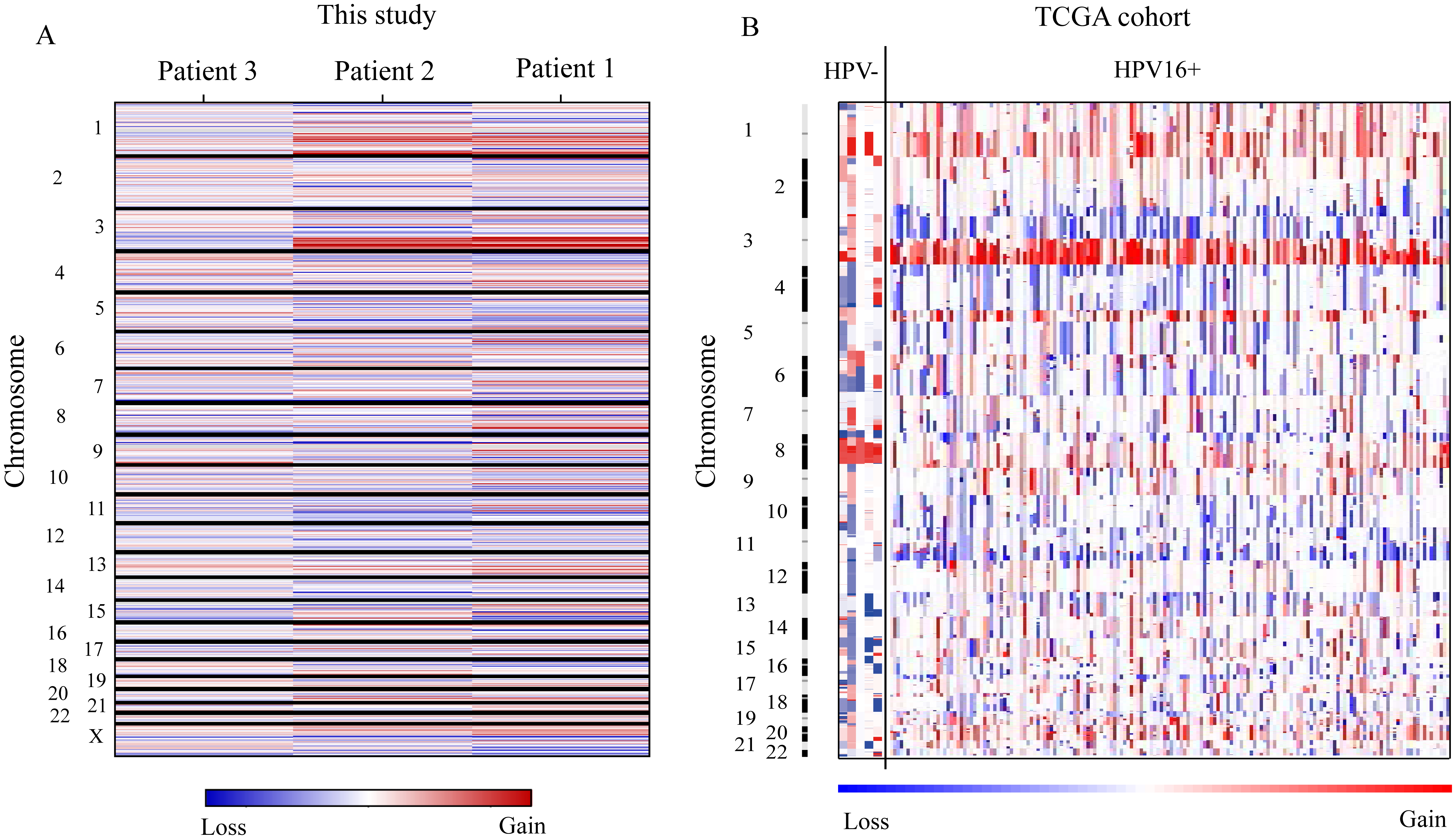
We found similar focal amplifications in this study and TCGA result, such as in 1q21, 1q25, 3q26, and 3q28. The most significant amplification peak located in 3q26 (Fig. 2A, 2B, Supplementary Fig. 1). The well-reported oncogene in 3q26 was PIK3CA, which was the most recurrent somatic mutated gene in cervical cancer [14] reported by TCGA study, meanwhile. And PI3K-AKT signaling activation was an important molecular classification of cervical cancer [14].
20345 expressed genes were detected by requiring that the FPKM value was greater than one. With the threshold of fold change
Different expressed genes (DEGs) in cervical cancer tissues. (A) Volcano plot of the differential expression analysis of genes; (B) Heatmap of unsupervised hierarchical clustering of DEGS in three samples; (C) The top 10 significantly upregulated and downregulated DEGs. Pathway and functional analysis of deregulated genes in cervical cancer by IPA (D) and GO (E) enrichment.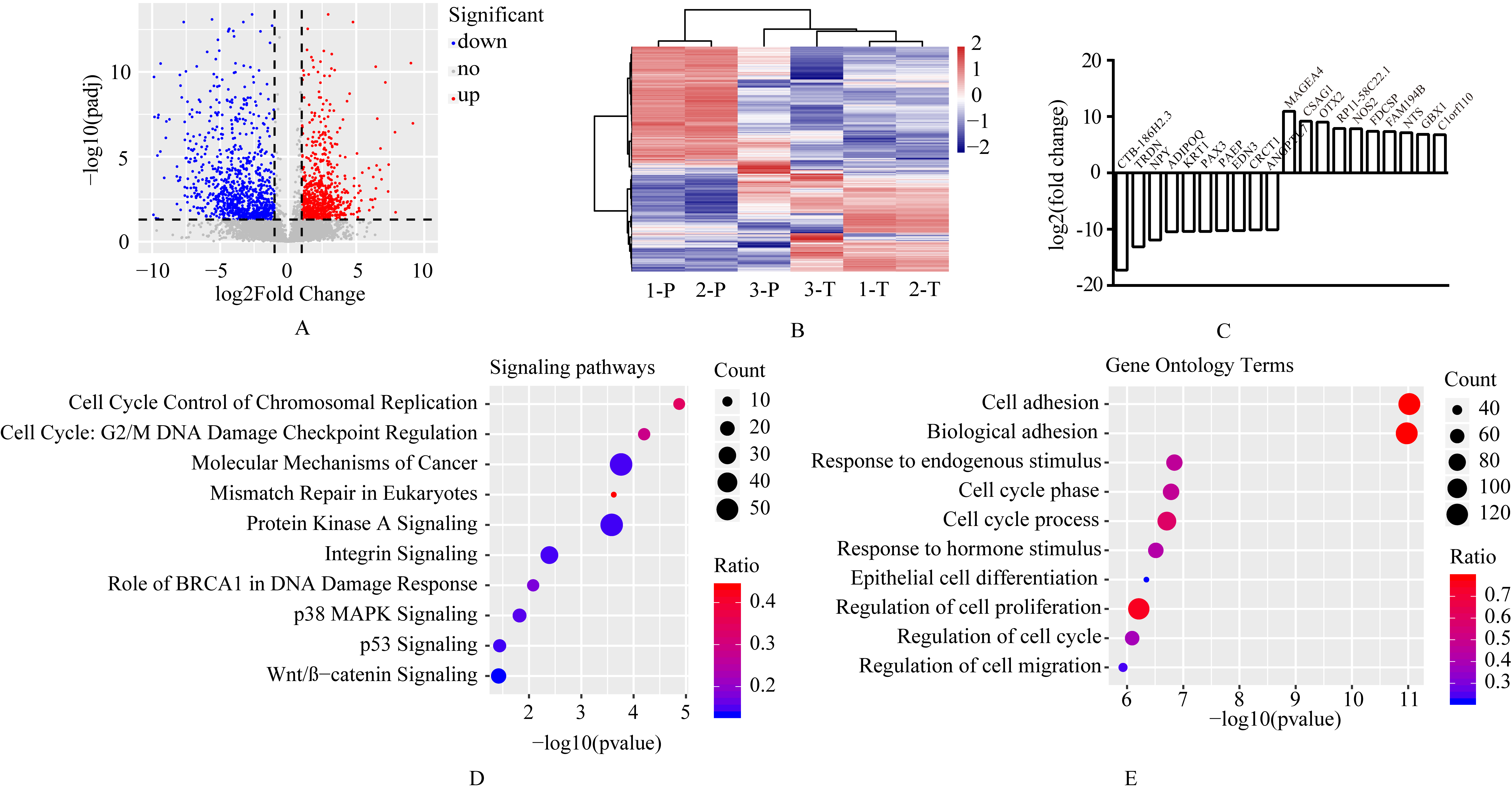
To better understand the biological function of these differentially expressed genes, signaling pathways (by IPA) and functions (by GO) enrichment analysis were performed. Figure 3D showed the top 10 cancer related signaling pathways including cell cycle control of chromosomal replication, cell cycle: G2/M DNA damage checkpoint regulation, and molecular mechanisms of cancer et al, ranked by
Notably, the most significant altered GO terms were related with adhesion function. Adhesion is a major factor affecting cell motility, and focal adhesions are essential in for cancer cell migration and invasion [18]. We also observed that cell cycle related pathway (cell cycle control of chromosomal replication, cell cycle: G2/M DNA damage checkpoint regulation) and GO terms (cell cycle phase, cell cycle phase, and regulation of cell cycle) were enriched. The role of cell cycle regulation in tumor development and progression has been extensively demonstrated [19, 20]. Among the identified GO terms, Endogenous stimuli may create a microenvironment for tumor growth, and responses to endogenous stimuli may induce changes in the microenvironment by self-secreted factors. These factors then produce changes in tumor metabolism, secretion, immunity, structure, and function. All of these processes affect tumor development and progression and can determine therapeutic responses [21].
Integrated analysis between CNVs and DEGs of tumors from patient 1 (A), patient 2 (B) and patient 3 (C).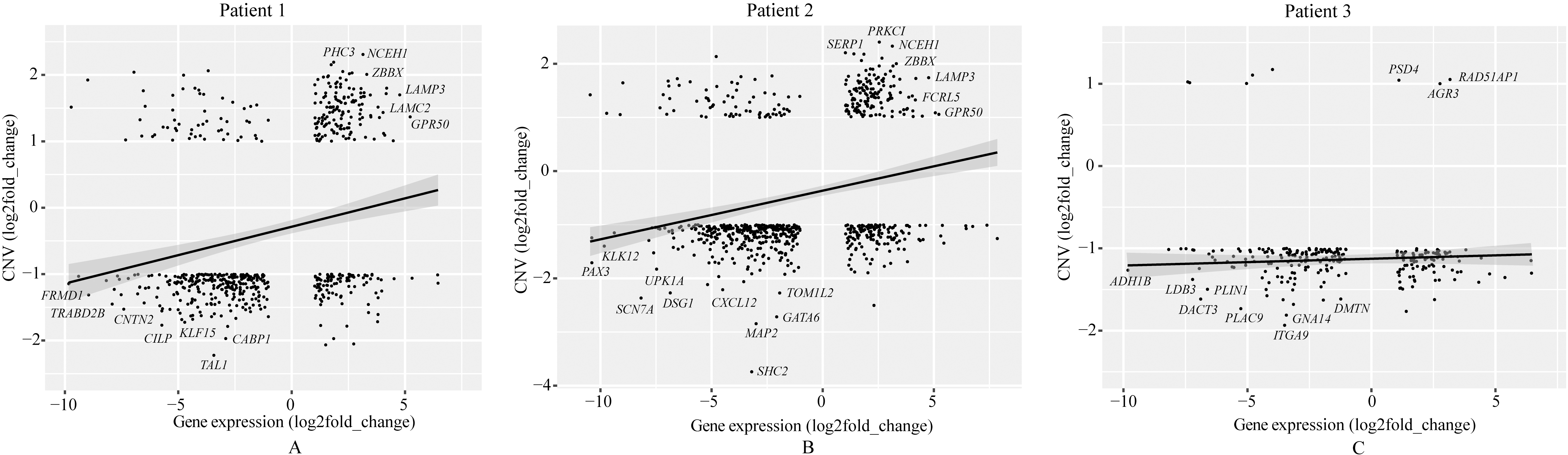
Based on the whole-exome sequencing, we identified a series of genes, including somatic mutated genes and CNVs. RNA sequencing identified up-regulated and down-regulated genes. Linear fitting analysis showed positive correlation between genes’ expression level and copy numbers in tumors of patient 1 and 2, but not in patient 3 (Fig. 4). Figure 5 presented statistics and compare of these positive-correlation genes among 3 patients. Tumors of patient 1 and patient 2 shared majority of genes (127 genes) with both up-expression and copy number gain. Among these 127 genes, for example, PHC3 amplification was reported as a possible cancer-driving genetic alteration in solid tumors by previous study [22]. While, few genes of sample 3 amplified. Tumors of 3 patients shared 45 genes with down-expression and copy number loss.
Statistics and compare of positive-correlation genes between expression level and gene copy number among 3 patients. (A) Genes with up-expression and copy number gain. (B) Genes with down-expression and copy number loss.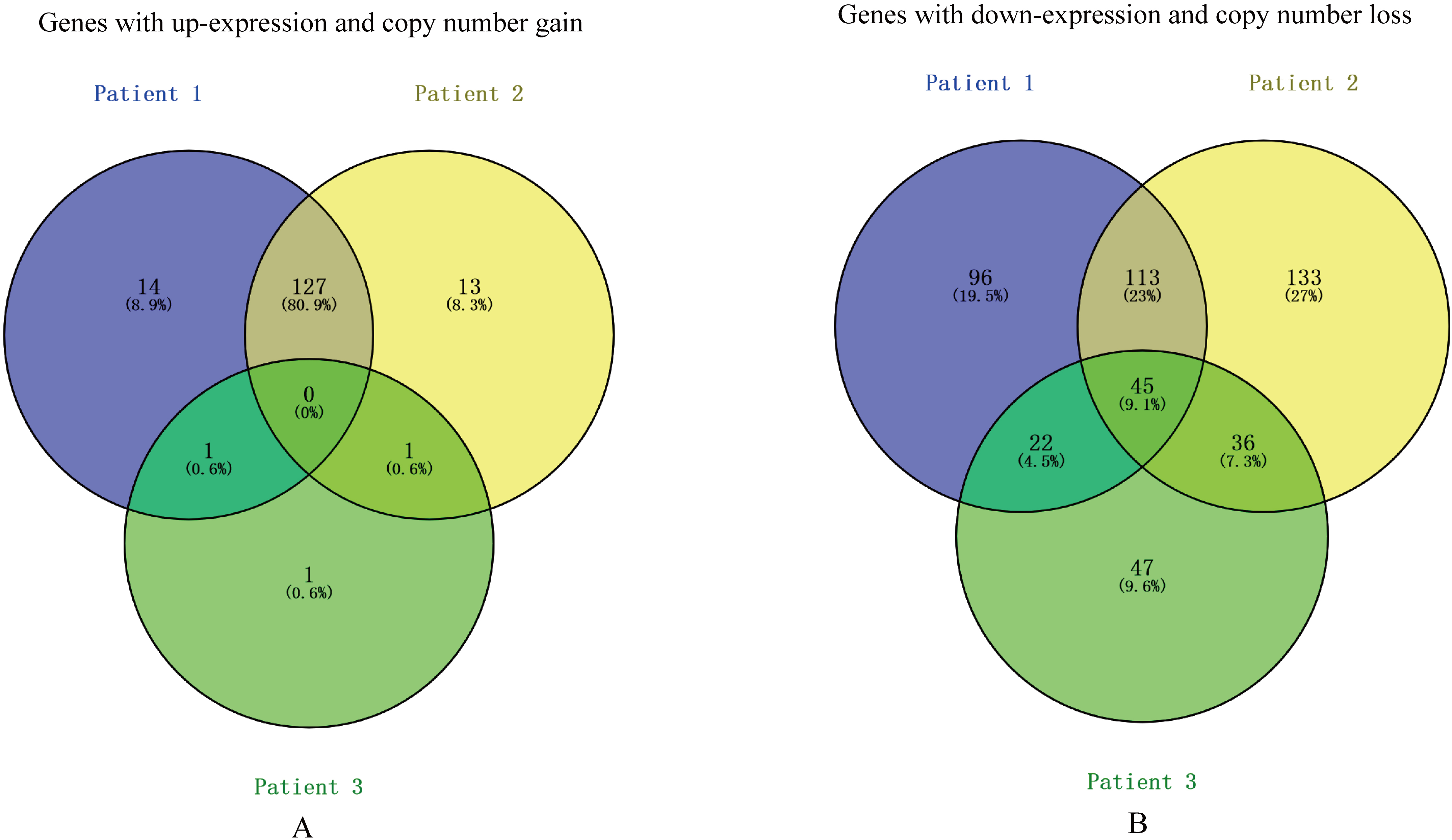
In this study, three cervical patients were recruited, and one of them (patient 3) was affected with HPV16. Whole-exome and RNA sequencing were employed to characterize the genome and transcriptome profiles of cervical cancer. We analyzed somatic mutated genes and CNVs based on whole-exome sequencing, and deregulated genes based on RNA sequencing. In addition, we analyzed whole-exome sequencing and Affymetrix SNP 6.0 arrays data of cervical cancer in TCGA cohort, as an independent validation group.
We found that somatic mutation profile, CNV heat map, and gene expression heat map were similar of tumors from two HPV-cervical cancer patients, and were distinct from tumors of HPV16+ patient. HPV16+ tumors had fewer somatic mutations than HPV-tumors, which was validated by TCGA results (Fig. 1). The following reasons may result in this phenomenon. Firstly, HPV16 was the driving factor of HPV16+ cervical cancer. While, HPV- tumors got more passenger mutations in the carcinogenic process, because of unclear driver factor. Previous studies reported the carcinogenicity of HPV results primarily from the activity of the oncoproteins E6 and E7, which impair growth regulatory pathways [23]. Integration of HPV DNA into the host genome is suggested to be a driver of the neoplastic process including loss of function of tumor suppressor genes, enhanced oncogene expression, loss of function of DNA repair genes, or other vital cellular functions [24]. Besides, adjacent normal tissue was infected with HPV16, which may result in genomic alterations not only in cervical tumor but also normal cervical tissue. Figure 3B indicated expression profile of normal cervical tissue of HPV16+ patient was little similar with cervical tumors. Somatic mutations were annotated taking adjacent normal tissue as control, so part of somatic mutations in normal tissue were filtered out in this process.
A combined analysis of whole-exome and RNA sequencing was performed. Linear fitting analysis showed positive correlation between gene expression level and copy numbers in HPV- cervical tumors (Fig. 4). Gene copy number alterations can reflect in gene expression level, which was well studied by researchers. But, expression level and copy number of many genes were inversed, because of gene expression level was affected by various factors such as promoter methylation, transcription factor binding, CNV et al. However, we found no positive correlation in HPV16+ cervical tumor, because fewer copy number gain genes were identified in its tumor.
In summary, a combined analysis of whole-exome and RNA sequencing was performed, and a series of mutations and DEGs was identified, which provided a molecular understanding of HPV associated cervical cancer. Although this is a preliminary study and suffers from having a small sample size, our results identified several genes involved in cervical cancer and provided novel insights into the pathogenesis of the malignancy.
Footnotes
Acknowledgments
This study was supported by the grants from the Natural Science Foundation of Fujian Province (grant no. 2014J01436), Jiangsu Provincial Project of Invigorating Health Care through Science, Technology and Education, Jiangsu Provincial Medical Youth Talent, the Project of Invigorating Health Care through Science, Technology and Education (grant no. QNRC2016621), and Major Projects of Special Development Funds in Zhangjiang National Independent Innovation Demonstration Zone, Shanghai (ZJ2017-ZD-012).
Conflict of interest
The authors declare no conflicts of interest.
Supplementary data
The supplementary files are available to download from