
Abstract
BACKGROUND:
Evidence indicates that inorganic arsenic (iAs) can directly damage cells and result in malignant transformation with unclear complicated mechanisms. In the present study, we aimed to explore the possible molecules, pathways and therapeutic agents by using bioinformatics methods.
METHODS:
Microarray-based data were retrieved and analyzed to screen the differentially expressed genes (DEGs) between iAs-treated lung cells and controls. Then, the functions of DEGs were annotated and the hub genes were filtrated. The key genes were selected from the hub genes through validation in The Cancer Genome Atlas (TCGA) cohorts. Possible drugs were predicted by using CMAP tool.
RESULTS:
Two datasets (GSE33520 and GSE36684) were retrieved, and 61 up-regulated and 228 down-regulated DEGs were screened out, which were enriched in various pathways, particularly metabolism-related pathways. Among the DEGs, four hub genes including MTIF2, ACOX1, CAV1, and MRPL17, which might affect lung cancer prognosis, were selected as the key genes. Interestingly, Quinostatin was predicted to be a potential agent reversing iAs-induced lung cell malignant transformation.
CONCLUSION:
The present study sheds novel insights into the mechanisms of iAs-induced lung cell malignant transformation and identified several potential small agents for iAs toxicity prevention and therapy.
Introduction
Lung cancer is the most commonly diagnosed cancer and the leading cause of cancer death in the world [1]. Epidemiological evidence indicates that cigarette smoking, alcohol consumption, ionizing radiation, and chronic inflammation are risk factors. Besides, genetic variations can also contribute to lung carcinoma risk [2]. Moreover, environmental carcinogens in the environment might have a long-term effect on human health. With the development of industry, heavy metal exposure has become a problem that can not be ignored.
Evidence showed that cadmium exposure may result in a number of cancers, including lung cancer [3]. A study conducted in Slovenia et al. showed that mercury exposure could increase lung cancer risk [4]. In addition, lead exposure might also have an association with an elevated risk of lung carcinoma [5]. Besides these heavy metals, Arsenic, a toxic metalloid presents in the earth’s crust, has attracted much attention. Inorganic arsenic (iAs) has been classified as a human carcinogen since 1980 [6]. People can be exposed to iAs through air, food and drinking water [7]. Evidence showed that long-term iAs exposure may have a correlation with a variety of diseases, such as cardiovascular disease, diabetes, and cancers [8]. Thus, iAs has become a serious public concern.
A meta-analysis indicated that people who exposed to iAs may have a 2.44-fold increased risk of lung cancer relative to that of non-exposed people [6]. Even a low-dose of iAs exposure could significantly increase lung cancer risk [9]. The molecular mechanisms of iAs carcinogenesis remain poorly understood. Several mechanisms have been presented, such as genotoxicity, altered cell proliferation, oxidative stress, changes to the epigenome, and disturbances of signal transduction pathways [10]. However, only a few studies have been devoted to the molecular mechanisms, with inconclusive results generated.
In recent years, high-throughput tools such as microarray and sequencing have been widely used to explore the gene variations in a variety of diseases, particularly carcinomas [11]. Exploring and mining massive data may help us screen important genes or pathways associated with the diseases. Hence, in the present study, we aimed to retrieve and analyze relevant microarray-based data and screen the differentially expressed genes (DEGs) that might play a role in iAs-induced bronchial epithelial cells damage. Next, the functions of the candidate genes were annotated. Then, hub genes among the DEGs were screened out and their roles in lung cancer were further assessed. Afterward, the agents that might rescue the cells from iAs-induced damage were predicted and evaluated.
Methods
Data source
A search in the Gene Expression Omnibus (GEO) database (
Screening of DEGs
To obtain the DEGs between iAs-treated lung cell group and the control group, the data of the selected gene expression profiles were analyzed by GEO2R [12]. The results were downloaded in text format, and the
If there were more than or equal to two datasets that met the inclusion criteria, the intersections of the up-regulated and down-regulated DEGs from each dataset, respectively, were obtained by using Venn analysis [13].
Functional enrichment analysis of DEGs
To evaluate the possible functions of the DEGs, the functional enrichment analyses of the DEGs, including gene ontology (GO) function analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were carried out by using Kobas database [14].
In the GO analysis, genes can be classified into hierarchical categories and the gene network can be presented according to biological processes [15]. An adjusted
The KEGG pathways database is a collection of graphical diagrams (pathway maps) for the biochemical pathways, whose efforts have been made to develop and improve the cross-species annotation procedure for linking genomes to the molecular networks through the KEGG Orthology system [16]. Fisher’s exact test and chi-square test was used to select the significant pathways. A
The most significant up-regulated and down-regulated DEGs in GSE33520 and GSE36684 (Top ten, iAs-treated cells versus controls)
The most significant up-regulated and down-regulated DEGs in GSE33520 and GSE36684 (Top ten, iAs-treated cells versus controls)
To find the possible hub genes/proteins that might play a key role in the biological process, the DEGs were analyzed by STRING and Cytoscape tools in order to predict the interaction relationship among them. A combined score of not less than 0.4 (median confidence score) was used as a threshold. The hub protein was selected based on its association with other proteins, which were sorted by the degree values in the network.
To explore the possible roles and prognostic values of candidate hub genes in lung cancer, two cohorts in the TCGA database were used for evaluation [17]. Thus, the data concerned two subgroups, namely, lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC), respectively. The expression levels and prognostic values were assessed for each hub gene. The genes whose expression levels affected the prognosis of lung cancer may be regarded as the key genes.
Identification of small molecules that reverse iAs-induced biological alterations
To predict the small agent for prevention of iAs-induced cell damage, the connectivity mapping (CMAP) database that contains whole genomic expression profiles for small active molecular inferences was used [18]. The DEGs were submitted to CMAP for analysis. Small molecular compounds with negative connectivity enrichment scores were evaluated and selected as potential therapeutic molecules.
Results
Identification of DEGs from the selected gene expression profiles
Two gene expression profiles, GSE33520 and GSE36684, were retrieved from the GEO database. GSE33520 contained seven specimens (three specimens of lung epithelial cells exposed to 2.5
Intersection of the up-regulated (a) and down-regulated (b) DEGs from GSE33520 and GSE36684, respectively. The intersection included 61 up-regulated and 228 down-regulated genes.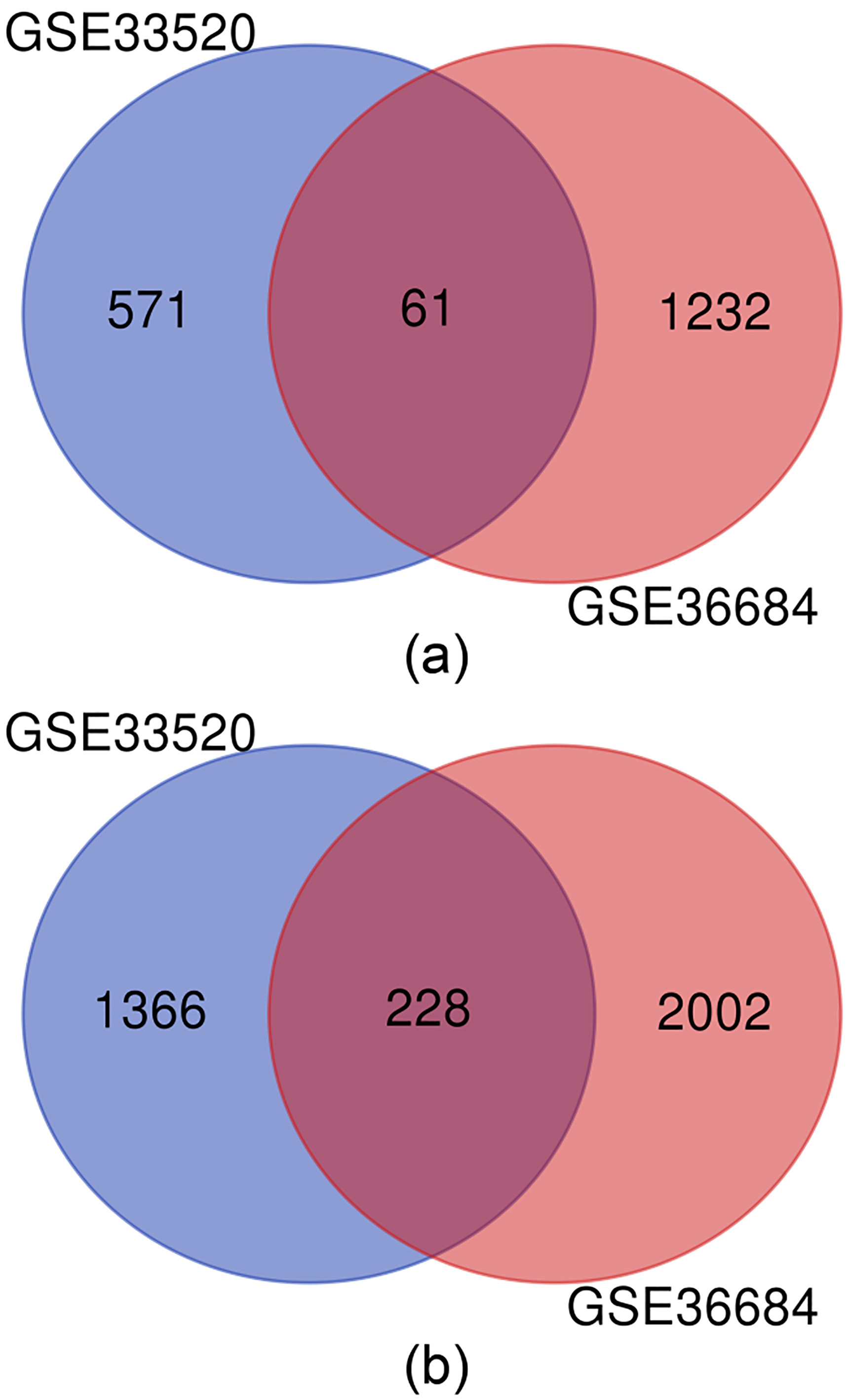
The DEGs were screened out from the comparison of the two groups, respectively. As a result, 632 up-regulated and 1594 down-regulated genes have been screened out from GSE33520, while 1293 up-regulated and 2230 down-regulated genes have been generated from GSE36684 (Table 1).
The top 10 terms of Go analysis (a) and KEGG enrichment analysis (b), respectively.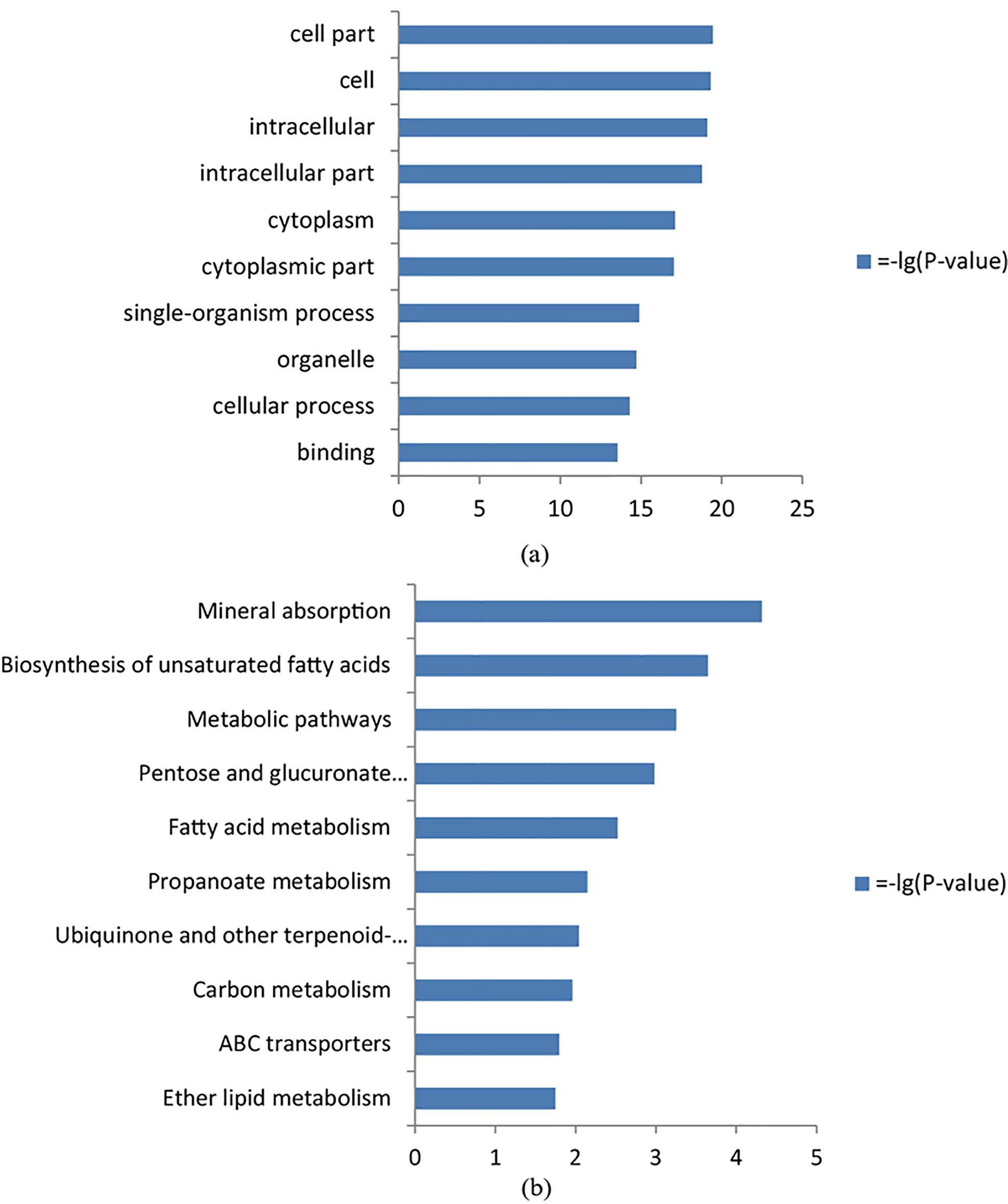
To get the shared DEGs in both datasets, Venn analysis was used to obtain the intersection of the DEGs profiles. As shown in Fig. 1, there were 61 up-regulated and 228 down-regulated genes in the intersections, respectively.
To explore the biological functions of the DEGs, they were clustered through GO analysis. GO analysis showed that the DEGs were enriched in 183 GO terms. The top items were cell part, intracellular part, cytoplasmic part, cellular process and binding (Fig. 2a).
KEGG pathway analysis showed that the DEGs were enriched in 23 pathways. The top items were Mineral absorption, Biosynthesis of unsaturated fatty acids, Metabolic pathways, Pentose and glucuronate interconversions, and Fatty acid metabolism (Fig. 2b).
PPI network construction
The DEGs in the intersection were submitted to STRING website for analysis. The nodes with a confidence of 0.4 were selected for constructing PPI networks.
In this network, 15 node genes/proteins, whose degrees were more than five, showed a strong association with other node proteins, respectively (Table 2, Fig. 3). Therefore, the screened hub genes (proteins) might play critical roles in iAs-induced lung epithelial cell malignant transformation.
The hub genes in the protein-protein interaction network and validation in TCGA cohorts
The hub genes in the protein-protein interaction network and validation in TCGA cohorts
LUAD: Lung Adenocarcinoma; LUSC: Lung Squamous Cell Carcinoma; Up (Down): The gene was up-regulated (down-regulated) in cancer tissues relative to normal tissues.
Protein-Protein Interaction network of the hub genes (proteins). The genes in the green modules were the top 15 hub genes sorted by the degree value.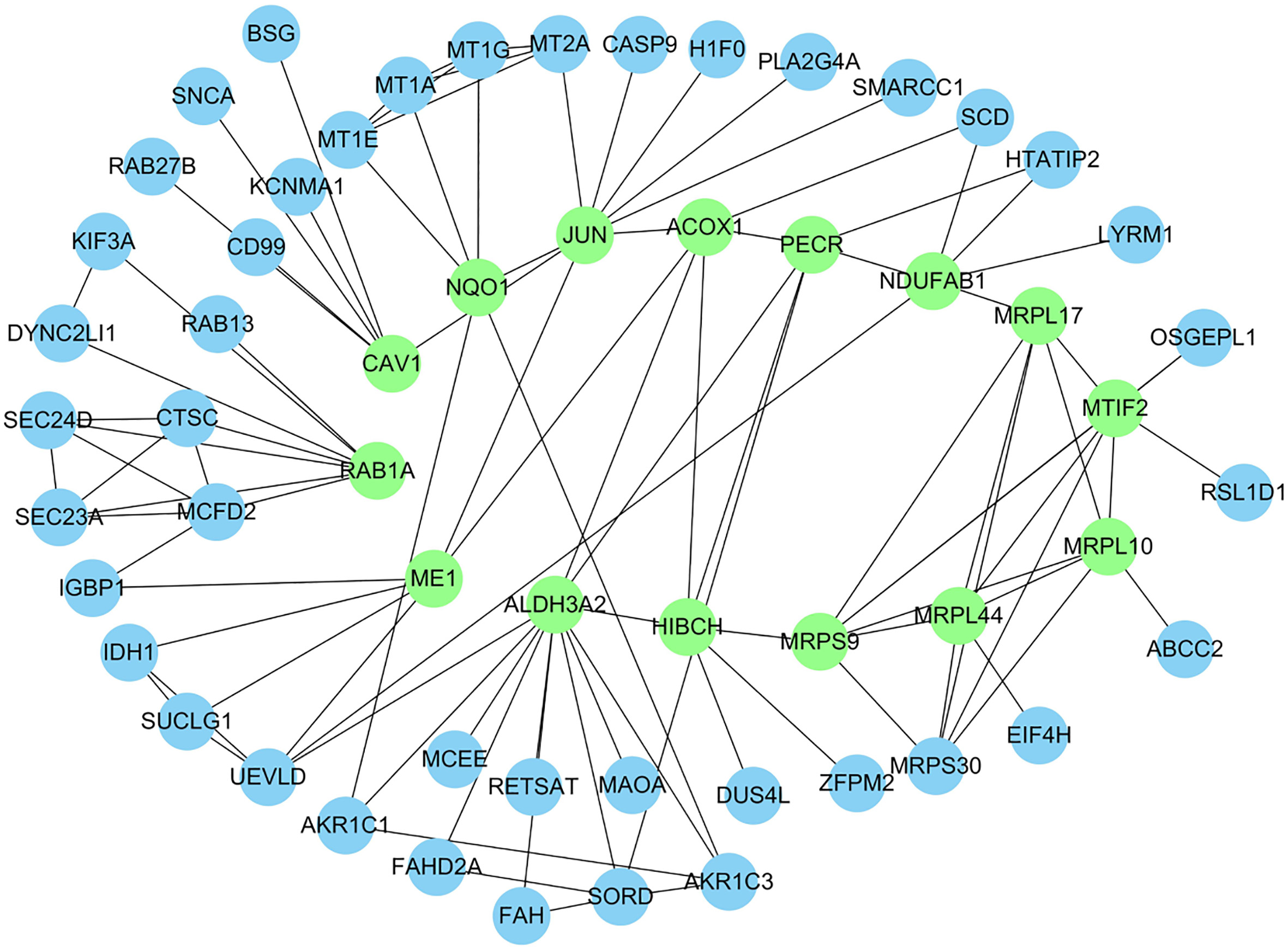
To explore the expressions and prognostic values of the hub genes in lung carcinoma, we validated these issues with two cohorts in TCGA.
In the TCGA database, the data regarding lung cancer were divided into two cohorts (LUAD and LUSC), respectively. The cohort of LUAD contained 515 cases and 59 normal controls; while that of LUSC comprised of 503 cases and 52 controls.
As shown in Table 2, most of the hub genes, except for ALDH3A2, were dysregulated in lung cancer tissues compared to normal tissues. Nevertheless, among the 15 hub genes, only four of them (MTIF2, ACOX1, CAV1, and MRPL17) might act as prognostic factors for LUAD. Notably, only one hub gene, CAV1, might have an association with LUSC prognosis (Fig. 4).
The expression levels (A) and prognostic values (B) of the four key genes in lung carcinoma based on TCGA cohorts. (a) MTIF2 in LUAD; (b) ACOX1 in LUAD; (c) MRPL17 in LUAD; (d) CAV1 in LUAD; (e) CAV1 in LUSC. 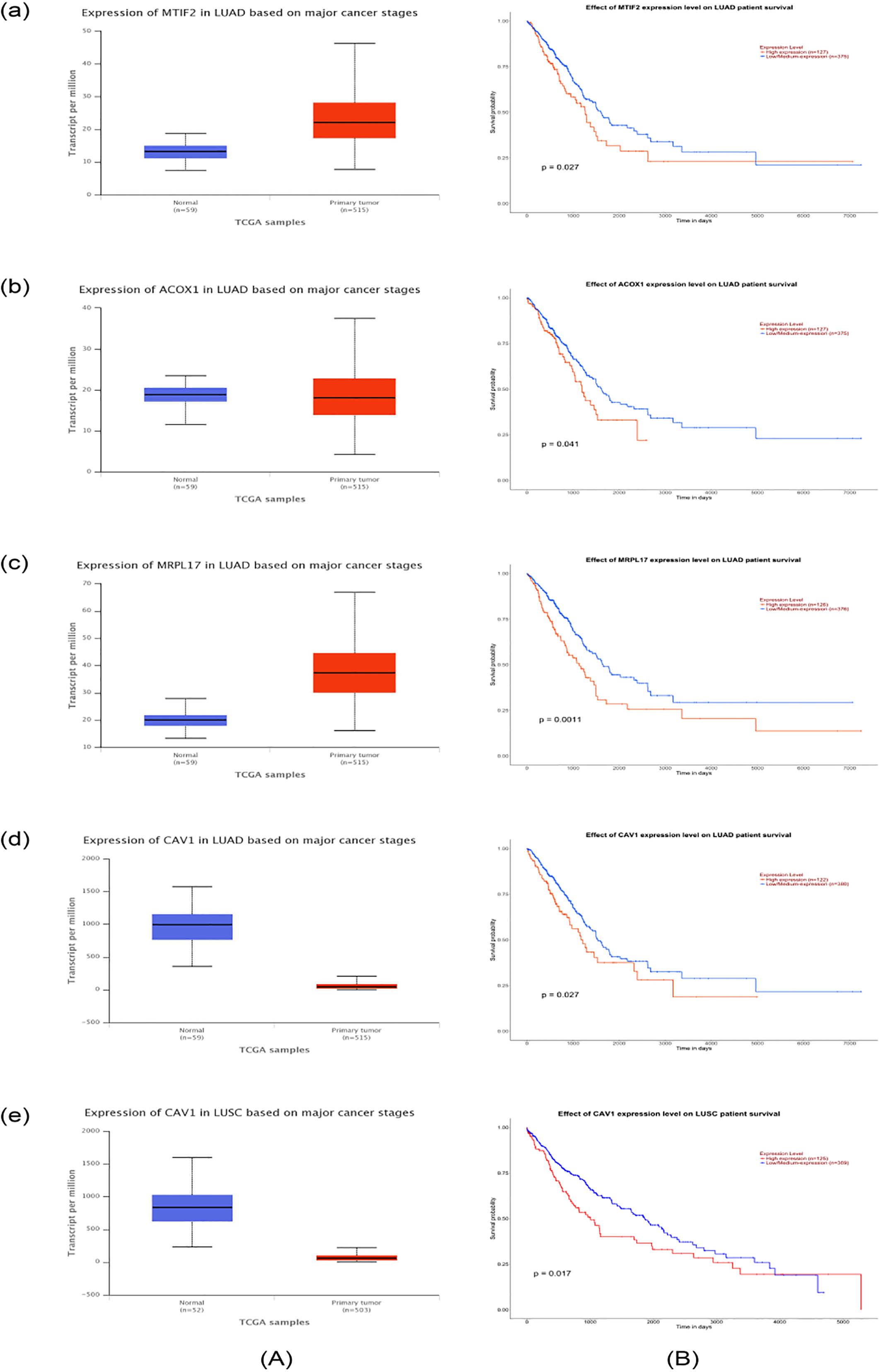
To further assess the prognostic value of these genes in lung cancer, we conducted Cox regression analyses by using the TIMER tool [19] that was also based on TCGA cohorts. As shown in Table 3, the results failed to show the four genes (MTIF2, ACOX1, CAV1, and MRPL17) as independent prognostic factors for lung carcinoma.
Multivariate analyses of several hub gene expressions and other clinical prognostic markers related to overall survival in lung cancer
HR: hazard ratio; CI: confidence interval; LUAD: Lung Adenocarcinoma; LUSC: Lung Squamous Cell Carcinoma.
To predict small molecules capable of reversing the iAs-induced lung cell malignant transformation, the DEGs were submitted to CMAP for analysis. The perturbagens from the CMAP were analyzed according to their permutated results,
Predicted small molecules in CMAP (top 5)
Predicted small molecules in CMAP (top 5)
As shown in this table, Quinostatin, Alcuronium Chloride, 5230742, Proscillaridin and Edrophonium Chloride might have a potential reversing the iAs-induced biological process, indicating that these agents need to be further validated as preventive or therapeutic drugs for iAs-associated lung cancer.
Heavy metal exposure has become a public concern for years. iAs exposure mainly through drinking water may result in cell damage or malignant transformation, thus increasing cancer risk in the exposed population. The mechanisms of iAs-induced cell malignant transformation have remained not clear. Previous reports have focused on the carcinogenic effects of the iAs metabolic products, such as MMA
GO analysis showed that the DEGs were enriched in 183 GO terms, such as cell part, intracellular part, cytoplasmic part, cellular process and binding, while pathway analysis showed that the DEGs were enriched in 23 pathways such as Mineral absorption, Biosynthesis of unsaturated fatty acids, Metabolic pathways, Pentose and glucuronate interconversions, and Fatty acid metabolism. The data indicated that the screened DEGs in the present study might be involved in multiple aspects of the biological process, and they might also be enriched in multiple signaling pathways. Of these pathways, cell metabolism may be worth noting because several top pathways were related to ‘metabolism’. This is consistent with previous evidence that metabolite of iAs may exert a toxic effect on lung cells [8].
Most of the hub genes were dysregulated in lung cancer tissues relative to normal tissues, indicating that these genes may be crucial in the iAs-induced tumorigenesis of lung cells. However, not all genes can affect the prognosis of lung cancer. After a validation analysis in TCGA cohorts, four genes, including MTIF2, ACOX1, CAV1, and MRPL17, were selected as prognostic factors for lung cancer, suggesting that the expressions of these genes may have an influence on the survival time of lung carcinoma patients. MTIF2 (Mitochondrial translation initiation factor 2) may initiate the translation of proteins encoded by the mitochondrial genome [21]. Little evidence has revealed its roles in cancer. Nevertheless, a recent report indicated that MTIF2 may be cell proliferation-related genes in glioma [22]. ACOX1 (acyl-CoA oxidase 1) is the first and rate-limiting enzyme in fatty acid
It is worth noting that both arsenic trioxide and NaAsO2 can often be used as Inorganic arsenic representatives in toxicological studies. The exposure of lung cells to Inorganic arsenic may usually result in two major events: cell apoptosis or cell malignant transformation. However, the mechanisms of these two Inorganic arsenic representatives on carcinogenesis are very complicated. Few investigations have been devoted to the comparison of the carcinogenesis mechanisms between them. Moreover, the goal of our paper was to find the possible key genes in iAs-related lung cancer by using bioinformatics methods. Thus,‘similarities and differences’ of arsenic trioxide and NaAsO2 can only be addressed in future studies.
To predict the small agent for reversing the iAs-induced cell malignant transformation, CMAP tool was utilized. As a result, a list of agents was predicted. The top molecules included Quinostatin, Alcuronium Chloride, 5230742, Proscillaridin, and Edrophonium Chloride. Among these molecules, Quinostatin had the highest negative enrichment score. Evidence indicated that Quinostatin can directly target the lipid-kinase activity of the catalytic subunits of class Ia PI3Ks, and inhibit mTOR network [30]. Quinostatin was also regarded as an inhibitor of cellular S6 phosphorylation [31]. In a recent study, Quinostatin has been predicted as a potential supplementary drug for treating pediatric acute lymphoblastic leukemia [32]. Therefore, since S6 phosphorylation, PI3K, mTOR signaling pathways might be involved in the iAs-induced cell transformation, Quinostatin might act as a potent agent for the prevention or treatment of iAs-associated lung carcinoma.
In conclusion, the present study provides preliminary research on the molecular mechanisms underlying iAs-induced bronchial epithelial cell transformation. High throughput data were analyzed to screen the DEGs by computational bioinformatics methods. The DEGs were mostly enriched in a variety of pathways, particularly cell metabolism-associated pathways. Then, several key hub genes that may play key roles in iAs-induced cell malignant transformation have been screened out. Using the CMAP tool, an mTOR inhibitor, Quinostatin, was predicted to have a potential reversing the iAs-induced carcinogenesis and development. However, future validation experiments are warranted to confirm the results.
Footnotes
Acknowledgments
This study was partially supported by the National Natural Science Foundation of China (No. 81672290).
Conflict of interest
The authors declare that they have no conflict of interest.