
Abstract
BACKGROUND:
Multidrug resistance of Hodgkin’s lymphoma (HL) often results in recurrence. Thus, we aimed to explore the underlying molecular mechanisms of multidrug resistance using bioinformatics strategies.
METHODS:
The gene expression profile was obtained from GEO database. Then, the differentially expressed genes were screened out, and their functional annotations were carried out. Then, gene-signal interaction network was constructed and Connectivity Map (CMAP) analysis was performed.
RESULTS:
A total of 1425 dysregulated genes were screened out, which were mainly enriched in biological items, such as small molecule metabolic, signal transduction, and cell apoptosis. Some survival-related pathways, such as MAPK pathways, apoptosis, and P53 pathway, and several hub genes, such as PRKCA, ACTN1, PIP5K1B, PRKACB, and JAK2, might play key roles in the development of multidrug resistance. Interestingly, felodipine was predicted to be a potential agent overcoming the multidrug resistance.
CONCLUSIONS:
The present study offered new insights into the molecular mechanisms of multidrug resistance and identified a series of important hub genes and small agents that might be critical for treatment of multidrug-resistant HL.
Keywords
Introduction
Lymphoma is a group of blood cell tumors that originate from lymphocytes, which are classified into two main categories, namely, Hodgkin’s lymphomas (HL) and non-Hodgkin lymphomas (NHL). Lymphomas in adolescents and young adults (AYA) accounting for 22% of all malignant tumors in patients aged 15–24 years (16% for HL and 6% for NHL), respectively [1].
The incidence of HL has gradually risen for decades. Radiotherapy and chemotherapy have been used as the major treatment methods for HL. However, the curable patients would inevitably encounter the risk of long-term toxicity during treatment [2]. Although many patients with HL may be cured by the current regimen of high-dose multi-agent chemotherapy, the treatment causes high risks of later pathologies including secondary malignancies [3]. Moreover, though more than 70% of HL patients are cured after treatment, there are about 30% of patients might experience recurrence [4]. This might be due to the occurrence of multidrug resistance that results from increased drug efflux by elevated expression of drug transporters [5].
The mechanisms of multidrug resistance are complicated. Several factors, such as multidrug resistance proteins and ATP-binding cassette families, are cell membrane transporters that can efflux chemotherapy agents from the cell cytoplasm [6]. To overcome the adverse effects of chemotherapy agents, various targeted therapeutic methods have been developed and several agents have shown more specific effects on cancer cells than chemotherapies. Nevertheless, although these agents have been very useful for cancer treatment, the subsequent presence of acquired resistance has reduced the efficacy of targeted therapies. In fact, the majority of the targeted methods concentrated on any certain molecular target, just ignoring that multidrug resistance has a relationship with aberrant expressions of multiple genes as well as pathways. Hence, it is urgent to explore the resistance-related multiple gene variations with more powerful genome-wide technologies, which may effectively clarify the nature of multidrug resistance and then help explore novel therapeutic strategies.
Microarray is a high-throughput tool for efficiently obtaining global gene expression profiles, which has been widely used for investigating the mechanisms underlying a growing body of diseases, including cancer [7].
In the present study, we aimed to explore the key genes or pathways involved in multidrug resistance of HL by using bioinformatics methods. Public microarray data were downloaded and differentially expressed genes (DEGs) among cells with different degrees of sensitivity to multiple drugs were identified. The functions of DEGs were then assessed by Gene Ontology (GO) annotation, and pathway enrichment. The hub genes were identified through gene-signal interaction network construction. Afterwards, possible agents for overcoming multidrug resistance of HL were predicted by using Connectivity Map (CMAP) database.
Methods
Data source
The gene expression profile of GSE26325 was downloaded from the GEO (Gene Expression Omnibus) public microarray database. This dataset was deposited by Staege et al. [8], in which a total of six specimens, involving 2 lowest sensitivity, 2 intermediate sensitivity, and 2 highest sensitivity HL cell lines, respectively, were available based on GPL96 [HG-U133A] Affymetrix Human Genome U133A Array.
Identification of differentially expressed genes (DEGs)
The dataset was submitted to Dchip software and GCBI platform (
Functional enrichment analysis of DEGs
The functional enrichment analyses of the DEGs, including gene ontology (GO) function analysis, pathway analysis, and pathway-net analysis were carried out.
GO analysis can organize genes into hierarchical categories and present the gene network according to biological processes, which becomes a tool for gene clustering, gene function prediction, evaluation of protein-protein interaction, disease gene prioritization and other applications [9].
Pathway analysis was used to determine the significant DEG-associated pathways according to Kyoto Encyclopedia of Genes and Genomes (KEGG). Fisher’s exact test and chi-squared test was used to select the significant pathways. The threshold of significance was based on P values and false discovery rate (FDR).
Pathway-net analysis was further conducted to select the most significant pathway of the DEGs, which is an interaction network of significant pathways containing DEGs as classified by KEGG database. Each pathway was marked by indegree, outdegree and total degree on the basis of its number of both upstream and downstream interaction pathways. A pathway with a high total degree indicates that this pathway might have a high tendency to regulate or be regulated by other pathways, and hence it might be a key pathway in the biological process.
The most significant up-regulated and down-regulated DEGs (Top 10, Lowest-sensitive cells versus highest-sensitive cells)
The most significant up-regulated and down-regulated DEGs (Top 10, Lowest-sensitive cells versus highest-sensitive cells)
The gene-signal network was conducted based on the screened DEGs, in which the gene interaction network was constructed. The hub gene was selected based on its association with other genes. The DEGs with more associations with other DEGs indicate their important roles in the Gene-signal interaction network.
Identification of small molecules that overcome multidrug resistance
The CMAP database (
Results
Identification of DEGs between Lowest-sensitive and highest-sensitive HL cell lines
The analysis has been conducted as shown in Fig. 1. Based on the public microarray dataset GSE26325, a total of 1425 DEGs between lowest-sensitive and highest-sensitive HL cell lines were screened out, in which 900 genes were upregulated and 525 genes were downregulated. The DEGs are shown to be clustered in Fig. 2 and the top ten DEGs were listed in Table 1.
The flow chart of the bioinformatics analysis.
Cluster analysis results for gene expression in lowest-sensitive and highest-sensitive groups. The expression values clustered in the red areas indicate up-regulation while the green areas indicate down-regulation.
GO Enrichment (a) and KEGG pathway analysis (b).
To investigate the altered biological functions of the DEGs, data were clustered through Gene Ontology (GO) analysis. A
Next, pathway analysis showed that the DEGs were enriched in 161 pathways. The main pathways included Metabolic pathways (104 genes), PI3K-Akt signaling pathway (43 genes), Epstein-Barr virus infection (33 genes), Pathways in cancer (41 genes), Transcriptional misregulation in cancer (31 genes) and so on.
The top 10 significantly altered GO terms and pathways were presented in Fig. 3.
To learn which pathways play a key role in the multidrug resistance of HL, we further conducted pathway-net analysis to screen the key pathways involved in this process. The analysis revealed that the top 5 pathways are MAPK signaling pathway (41 degree), Apoptosis (31 degree), Pathways in cancer (28 degree), Cell cycle (23 degree), and p53 signaling pathway (18 degree) (Fig. 4). The results showed that the key pathways are important pathways involved in cancer progression.
Gene signal-network
A signal-net analysis was performed to screen the hub genes that might play key roles in the multidrug resistance of HL. With signal-net, important DEGs were screened out (Table 2 and Fig. 5). The results showed that the genes with top 5 betweenness were PRKCA, ACTN1, PIP5K1B, PRKACB and JAK2.
Results of the pathway-net analysis.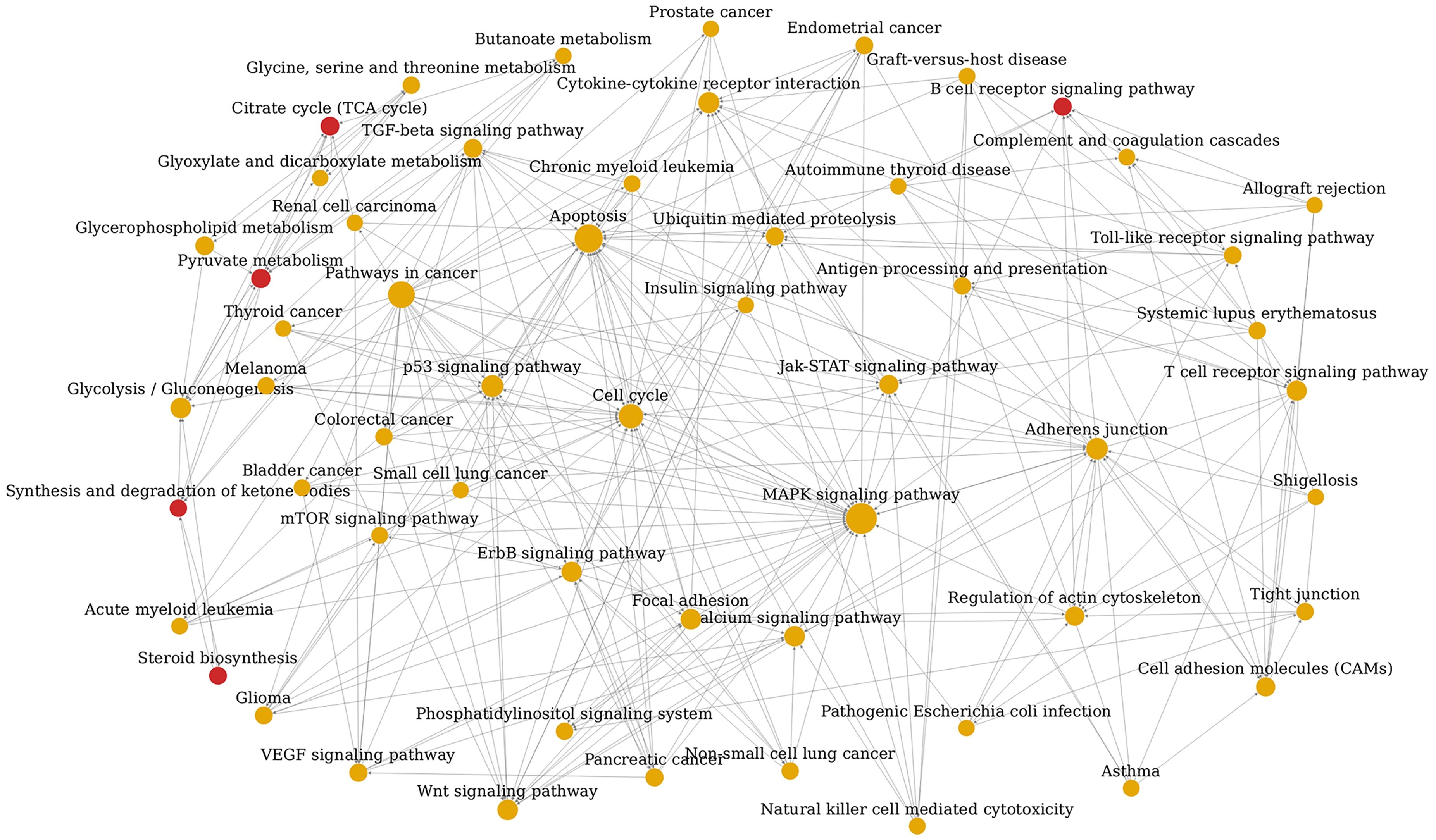
In order to screen out small molecule drugs that might be capable of reversing the gene alterations, the DEGs were submitted to CMAP, a bioinformatic tool that shows functional connections between small agents and gene expression signatures of diseases, for analysis. The perturbagens from the CMAP were analyzed according to their permutated results,
Discussion
In the present study, we used bioinformatic methods to explore the possible molecular mechanisms underlying the multidrug resistance of HL. The results showed that a total of 1425 DEGs were screened out. The GO analysis indicated that these genes were mainly enriched in items such as small molecule metabolic, signal transduction, and cell apoptosis. Then, pathway analysis revealed that some survival-related pathways such as MAPK pathways and P53 pathway may play critical roles in the multidrug resistance of HL. Interestingly, several genes such as PRKCA, ACTN1, PIP5K1B, PRKACB and JAK2, may be the hub genes that might play key roles in this process. Subsequently, using the CMAP tool, a small molecule, felodipine, was predicted to be a potential agent overcoming the multidrug resistance of HL.
Previously, several studies have tried to explore the mechanisms of multidrug resistance of HL. For example, mTOR has been thought to be an important pathway involved in HL multidrug resistance, and thus Everolimus, an inhibitor of mTOR, has been considered as a potential treatment drug for reversing the multidrug resistance [10]. Besides, over-expression of Glutathione-S-transferases has a correlation with multidrug resistance of HL [11]. Moreover, IL-4 and IL-13-mediated STAT6 activation has also been suggested to play a role in multidrug resistance of HL [3]. However, only any certain pathway was considered in each of these published studies. Since HL is a disorder characterized by multi-steps and multiple involved genes and pathways, use of bioinformatics methods for analyzing high throughput data may objectively help find key genes and pathways. To our knowledge, we for the first time used bioinformatics methods looking for the underlying molecular mechanisms of HL multidrug resistance.
Through GO analysis and pathway analysis, we found that some items may have a close association with HL multidrug resistance. For instance, signaling transduction, cell apoptosis, and small molecule metabolic process may be the key mechanisms. The pathway-net analysis further indicated that MAPK and P53 pathway may play critical roles during this process. Evidence showed that MAPK p38 pathway was involved in the multidrug resistance of oral carcinoma [12]. Although this signaling pathway has been studied for more than 25 years, its intricate dynamic control and plasticity regarding drug resistance are still unclear [13]. Thus, MAPK signaling pathway might play a key role in the multidrug resistance of HL. P53 is a tumor suppressor that has been indicated to be mutated in a variety of cancers and has been suggested to have a relationship with drug resistance [14]. Reports showed that up-regulated expression of P53 reversed chemoresistance of osteosarcoma cell lines [15]. In lung cancer, mutant P53 confers chemoresistance to cisplatin through upregulation of Nrf2 expression [16]. Hence, signaling pathways such as MAPK and P53 may be the key pathways involved in the multidrug resistance of HL.
Hub genes in the gene-signal network (top 5)
Predicted therapeutic small molecule agents with potential abilities to overcome multidrug resistance (top 10)
Gene-signal interaction network diagram of the 5 node genes (proteins) including PRKCA, ACTN1, PIP5K1B, PRKACB and JAK2, which showing more association with other node proteins, indicating they are hub genes.
Through gene-signal network construction, a series of hub genes (proteins) have been shown to form a local network, including PRKCA, ACTN1, PIP5K1B, PRKACB and JAK2, of which, several genes have been implied to be related to cancer development or drug resistance. Over-expression of PRKCA may confer chemoresistance in breast cancer [17] and ovarian cancer [18], and its inhibition results in reversion of the resistance. ACTNs are known to crosslink actin filaments at focal adhesions in migrating cells and thus may play a role in cancer cell motility and invasion [19]. As a key regulator of the actin cytoskeleton, its up-regulation in melanoma may promote the cell invasive abilities [20]. PIP5K1B is an enzyme functionally linked to actin cytoskeleton dynamics that phosphorylates phosphatidylinositol 4-phosphate [PI(4)P] to generate phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2]. Knockdown of PIP5K1B in cells results in abnormal actin cytoskeleton remodeling [21]. Thus, aberrant expression of PIP5K1B has been suggested to play a role in the progression of lung adenocarcinoma [22]. PRKACB is a member of the Ser/Thr protein kinase family, which was also a key effector of the cAMP/PKA-induced signaling transduction involved in various cellular processes, such as cell proliferation and differentiation. Therefore, downregulation of PRKACB might promote lung cancer development [23]. JAK2 is a major component of JAK2/STAT3 signaling pathway, which participates in autophagy-related drug resistance of bladder cancer cells [24]. Inhibition of JAK2/STAT3 could sensitize platinum-resistant cells to carboplatin [25]. Collectively, the above evidence supported the notion that the hub genes might play important roles in the multidrug resistance of HL. Nevertheless, it is still unknown whether these above hub genes can be used as targets for the drug resistance reversion. The data of the present study are preliminary findings. Future evaluations for the potential roles of the hub genes are required.
CMAP is an online tool that profiled human cancer cell lines exposed to a library of anticancer compounds with the goal of connecting cancer with underlying genes and potential treatments [26]. Through this tool, a list of agents that might reverse the DEGs profiles were screened out, of which felodipine, a member of the 1,4-dihydropyridine class of calcium channel blockers, had the highest negative enrichment score and thus it might have the potential to reverse the multidrug resistance of HL. Felodipine has been used to treat hypertension and angina and it has no negative inotropic effects at clinically administered doses [27]. Reports showed that calcium antagonists including felodipine can reverse ABCG2/BCRP-mediated drug resistance and transport in SN-38-resistant HeLa cells [28]. Moreover, in a study regarding cholangiocarcinoma, computational bioinformatics analysis identified that felodipine could induce inverse gene changes to the signature using the CMAP as analyzed in the present study. Then, in their further validation experiment, felodipine was indicated to have a synergistic effect with gemcitabine on cancer cells in vitro and in vivo [29]. Thus, the evidence led us to hypothesize that felodipine might be an effective agent overcoming multidrug resistance of HL.
In conclusion, the study provides preliminary investigation for the mechanisms underlying the multidrug resistance of HL. The DEGs of the multidrug resistance of HL were screened out by computational bioinformatics methods based on microarray data. Then, the aberrant pathways involved in this process were identified. Afterwards, several key hub genes were selected as potential targets for reversing the multidrug resistance of HL. Through the CMAP tool, agents that might have the potential to reverse the drug resistance have been predicted and analyzed. The results of the present study may give a valuable clue for both the basic research and clinical treatment of drug resistant HL. However, future validation experiments are warranted to test the findings.