
Abstract
Melanoma treatment with the BRAF V600E inhibitor vemurafenib provides therapeutic benefits but the common emergence of drug resistance remains a challenge. To define molecular mechanisms of vemurafenib resistance, we generated A375-R, WM35-R cell lines resistant to vemurafenib and show that the phosphorylated (p)-STAT3 was upregulated in these cells in vitro and in vivo. In particular, activation of the Signal-transducer-and-activator-of-transcription 3 (STAT3) pathway was associated with vemurafenib resistance. Inhibition of this pathway with STAT3-specific siRNA (shRNA) sensitized A375-R, WM35-R cells to vemurafenib and induced apoptosis in vitro and in vivo. Moreover, targeting STAT3 induced expression of miR-579-3p and elicited resistance to vemurafenib. However, targeting microRNA (miR)-579-3p with anti-miR-579-3p reversed the resistance to vemurafenib. Together, these results indicated that STAT3-mediated downexpression of miR-579-3p caused resistance to vemurafenib. Our findings suggest novel approaches to overcome resistance to vemurafenib by combining vemurafenib with STAT3 sliencing or miR-579-3p overexpression.
Introduction
Melanoma is the most aggressive form of skin cancer. Patients with metastatic disease have a median survival of less than one year, and outcomes are not improved with chemotherapy [1]. The immunotherapy is currently changing the landscape of oncology. Nowadays the standard of care in metastatic or unresectable melanoma patients include immunomodulating modalities such as anti-PD-1 drugs (nivolumab, pembrolizumab) and anti-CTLA-4 antibody ipilimumab. Ipilimumab is the only approved fully human monoclonal antibody that targets cytotoxic T-lymphocyte antigen-4 (CTLA-4) [2]. Toxicity of ipilimumab occurs in about 50–70% of patients, with 10–20% being serious, mostly immune-related adverse events [3]. Both anti-PD1 and anti-PD-L1 antibodies are now in clinical trials. Nivolumab was the first anti-PD1 antibody to be tested in a phase I study as a single agent in several tumour types, including melanoma [4]. Grade 3 toxicity consisted of CD4 lymphopaenia, fatigue and musculoskeletal problems. The toxicity profile of anti-PD1 appears to be better compared with ipilimumab. However, validated predictive biomarkers are still lacking.
B-type RAF proto-oncogene (BRAF) somatic mutations that render BRAF constitutively active are observed in 50%–60% of malignant melanomas [5]. Thus, BRAF inhibitors have recently shown promise for the treatment of metastatic melanoma harboring such BRAF mutations [6]. PLX4032 (vemurafenib), a selective RAF inhibitor, showed an unprecedented antitumor response rate in patients with BRAF(V600E) [7] and conferred an overall survival benefit in a pivotal phase 3 study [8]. Unfortunately, most patients rapidly acquire resistance to vemurafenib [9].In addition, although RAF inhibitor treatment has produced clinical responses in some patients, a subset of tumors is resistant to this agent, indicating the presence of intrinsic resistance or sensitivity to RAF inhibitor, highlighting the urgent need for new treatment strategies of BRAF(V600E)-induced melanoma. Besides BRAF inhibitors, most anticancer drugs have the problem of drug resistance, which limits their effectiveness [10].
Signal-transducer-and-activator-of-transcription 3 (STAT3), a transcription factor involved in cytokine signaling, participates in the regulation of cell cycle, apoptosis, cell invasion, and angiogenesis [11]. Targeting STAT3 signaling is an attractive therapeutic approach for most types of human cancers with constitutively activated STAT3, including melanoma [12, 13, 14]. Recently, it has found that the cell viability studies in B16F10 mouse melanoma cells have shown that the co-delivery of curcumin and STAT3 siRNA significantly inhibited the cancer cell growth compared with either liposomal curcumin or STAT3 siRNA alone [15]. Bid et al. [16] has reported that dual targeting BRAF(V600E) signaling and STAT3 signaling may be effective in selumetinib-resistant tumors or may retard or prevent onset of resistance in an in vivo model of childhood astrocytoma. A recent study has reported that STAT3 signaling is induced in BRAF-inhibitor-resistant cells, which made expression of phosphorylated Signal Transducer and Activator of Transcription 3 (pSTAT3) increase in BRAF-resistant melanoma cells [17]. Liu et al. [18] showed that activation of STAT3 induced resistance to vemurafenib in melanoma cells. In addition, STAT3 silencing inhibited the growth of melanoma cells with acquired resistance to vemurafenib. Furthermore, treatment with the STAT3 inhibitor, WP1066, resulted in growth inhibition in both vemurafenib-sensitive and -resistant melanoma cells. We therefore suggested that STAT3 inhibitor could enhance the sensitivity of RAF inhibitor to melanoma.
MicroRNAs (miRNAs) are small noncoding RNAs that modulate gene expression by mRNA silencing or degradation, which usually have pleiotropic effects because of their ability to target simultaneously multiple mRNAs. Changes in miRNAs expression levels are known to play a key-role in various human cancers. Recently, microRNAs have been reported to play an important role in inducing resistance to anti-cancer drugs, and specific microRNA alterations occur selectively in cancer cells, rendering these cells resistant to various chemotherapeutic agents [19, 20]. Studies previously showed that expression levels of miR-579-3p decrease from nevi to stage III/IV melanoma samples and even further in cell lines resistant to BRAF/MEK inhibitors. And miR-579-3p ectopic expression impairs the establishment of drug resistance in human melanoma cells [21].
In this paper we identify a mechanism of drug resistance in BRAF mutated melanoma centered around a poorly characterized miRNA, miR-579-3p.
Materials and methods
Chemicals and reagents
Antibodies against p-STAT3 (Y705), BRAF, STAT3 and b-actin were obtained from Santa Cruz Biotechnology (Shanghai, China). Vemurafenib was obtained from Selleck Chemicals (Hangzhou, China). TaqMan probes for miR-579-3p was purchased from Applied Biosystems; RNU48 (RNU48: 001006), a component of the spliceosome upon which splicing of pre-mRNA occurs, was from Life Technologies, Foster City, CA, USA.
Cell culture of melanoma cell lines
The human BRAFV600E mutated melanoma cell lines A375, WM35 were maintained in high-glucose RPMI 1640 supplemented with 5% fetal bovine serum, the cells were cultured at 37
miRNA transfection
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was used for the transfection of anti-miR-579-3p. Forty-eight hours after transfection, cells were treated with PLX4032 for response and apoptosis analysis and real-time PCR.
siRNA Transfection
Twenty nM siRNA (synthesized by biomers.net Germany, Shanghai, China) against STAT3 (sense: aacuucagacccgucaacaaa-dTdT; antisense: uuuguugacg ggucugaag-dTdT) were reversely transfected using the riboxx FECT (riboxx Life Sciences) on 96 well plates. Therefore, the riboxxFECT (1:25) and siRNA were separately diluted using Opti-MEM (Life technologies) and the solutions were gently mixed in a 1:1 ratio. The solutionwas incubated for 15 min and subsequently, 50
Construct stable short hairpin RNA (shRNA)-expressing cell lines
Silencing of STAT3 in A375-R and WM35-R cell lines was achieved using lentiviral infection. Lentiviral plasmid vectors containing-control and STAT3 short hairpin RNA (shRNA; Open Biosystems) were co-transfected with packaging vectors and the lentivirus was produced in 293T cells via calcium transfection as the standand methods. STAT3 shRNA-containing vectors were titrated, and A375-R and WM35-R was infected with 25
Cell viability assay
Cells were seeded in 96 well plates overnight and treated with increasing concentrations of drugs or DMSO (vehicle). The DMSO concentrations were maintained at 0.02% in all wells. After 48 h incubation, cell viability was determined using Cell Titer Blue Cell Viability Assay (Promega, Madison, WI, USA). Median inhibitory concentration (IC50) values were determined using in house software (DIVISA) and plotted in dose-response curves.
FCM analysis of cell apoptosis
The STAT3 siRNA and control siRNA transfected A375-R and WM35-R cells were subsequently treated with up to 10
Western blot
RIPA buffer was used for total protein extraction. Twenty
Real-time PCR
Total RNAs were extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed using the Taqman microRNA Kit (Applied Biosystem). The Taqman miRNA Assay was used for quantitative PCR. Ct values were employed for quantification of transcripts. MiRNA expression was normalized to the values of RNU48.
Immunofluorescence staining and confocal microscopy
Immunofluorescence was performed using standard techniques. Briefly, cells grown on coverslip in a six-well plate were fixed with 4% paraformaldehyde in PBS. Cells were rinsed with 1
STAT3 Knockdown sensitizes melanoma cells towards Vemurafenib treatment in vivo
This study was carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals of the USA National Institutes of Health, and the study protocol was approved by the Committee on the Ethics of Animal Experiments of The Affiliated Hospital of Qingdao University. For the in vivo tumor growth assays, 1
Immunohistochemistry
Staining of pSTAT3 on tissue sections by immunohistochemistry (IHC) was carried out by using standard techniques. pSTAT3 was detected with 3, 3’diaminobenzidine (DAB). Sections were counterstained with Harris hematoxylin.
TUNEL staining
Fresh-frozen sections of tumor tissues from therapy experiments were stained by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL; green; Promega, Madison, WI, USA) and counterstained with Hoechst 1:10,000. An apoptotic body was represented by green fluorescence. To quantify apoptotic cells, TUNEL-positive cells were calculated in 10 random fields at 200
Statistical analysis
For cell culture and animal experiments, data were analysed using GraphPad Prism software for Windows 7 (version 4; GraphPad Software, San Diego, CA, USA) and the two-tailed Student’s
Results
pSTAT3 expression is increased in vemurafenib-resistant melanoma cell lines in vitro
To unravel the functional relevance of STAT3 expression on BRAF V600E inhibitor (BRAFi) resistance development we generated BRAFi resistant melanoma cell lines (A375-R and WM35-R) by continuous treatment with vemurafenib. The in vitro vemurafenib dose-response curves for the parental A375-S, WM35-S and the vemurafenib-resistant A375-R and WM35-R cells shown in Fig. 1A. From the survival curves, we computed the respective IC
Western blot analyses of fractionated cell lysates confirmed that the resistance acquired cell lines A375-R, WM35-R exhibit significantly increased pSTAT3 expression compared to the parental A375-S and WM35-S cells (Fig. 1B). To further support that pSTAT3 was activated in the BRAFi resistant melanoma cell lines, we used immunofluorescence staining and confocal microscopy. As shown in Fig. 1C, prominent expression of pSTAT3 in vemurafenib resistant melanoma A375-R, WM35-R cells in comparison to the sensitive parental cells (Fig. 1C), indicating that vemurafenib resistant melanoma cells were with high pSTAT3 expression.
The STAT3 activity confers BRAF inhibitor resistance in BRAF mutant melanoma cells. A, Proliferation of A375-S, WM35-S and the vemurafenib-resistant A375-R and WM35-R cells in the presence of PLX4032 for 24 hs. IC50 values are means (
Targeting STAT3 restores sensitivity of melanoma cells to PLX4032 treatment. A, Western blotting confirmed that STAT3 was inhibited with STAT3-specific siRNA. B, Knockdown of STAT3 sensitized A375-R or A375-S cells to PLX4032 by cell viability. C, Knockdown of STAT3 sensitized WM35-R or WM35-S cells to PLX4032 by cell viability. D, The TUNEL assay in A375-R cells was performed using the One-Step TUNEL Apoptosis Assay Kit. E, The TUNEL assay in WM35-R cells was performed using the One-Step TUNEL Apoptosis Assay Kit. Images were captured by fluorescence microscopy. The green color is indicative of TUNEL-positive cells. *
Targeting STAT3 increased sensitivity of melanoma cells to PLX4032 treatment through miR-579-3p upregulation. A, miR-579-3p expression was determined by qRT-PCR in A375-R/S and WM35-R/S cells; B and C, miR-579-3p expression was determined by qRT-PCR in A375-R and WM35-R cells following different treatment. D and E, Cell apoptosis was detected by TUNEL staining in A375-R and WM35-R cells following different treatment. *
STAT3 inhibit sensitized A375-R or WM35-R cell to vemurafenib in vivo. Tumor growth in nude mice transplanted with A375-R cells (A) or WM35-R cells (B) treated with vemurafenib or STAT3 shRNA or their combination compared to that in untreated mice. Tumor volume of the transplanted mice was measured for a maximum of 40 days. C, Representative in vivo luciferase images of A375-R xenograft nude mice at day 40 after vemurafenib treatment. D, Representative in vivo luciferase images of WM35-R xenograft nude mice at day 40 after vemurafenib treatment. E, Immunohistochemical staining for p-STAT3 (Y705) with A375-R cells transplanted tumor tissue specimens; F, Immunohistochemical staining for p-STAT3 (Y705) with WM35-R cells transplanted tumor tissue specimens. G, miR-579-3p expression was determined by qRT-PCR in A375-R cells transplanted tumor tissue specimens; H, miR-579-3p expression was determined by qRT-PCR in WM35-R cells transplanted tumor tissue specimens.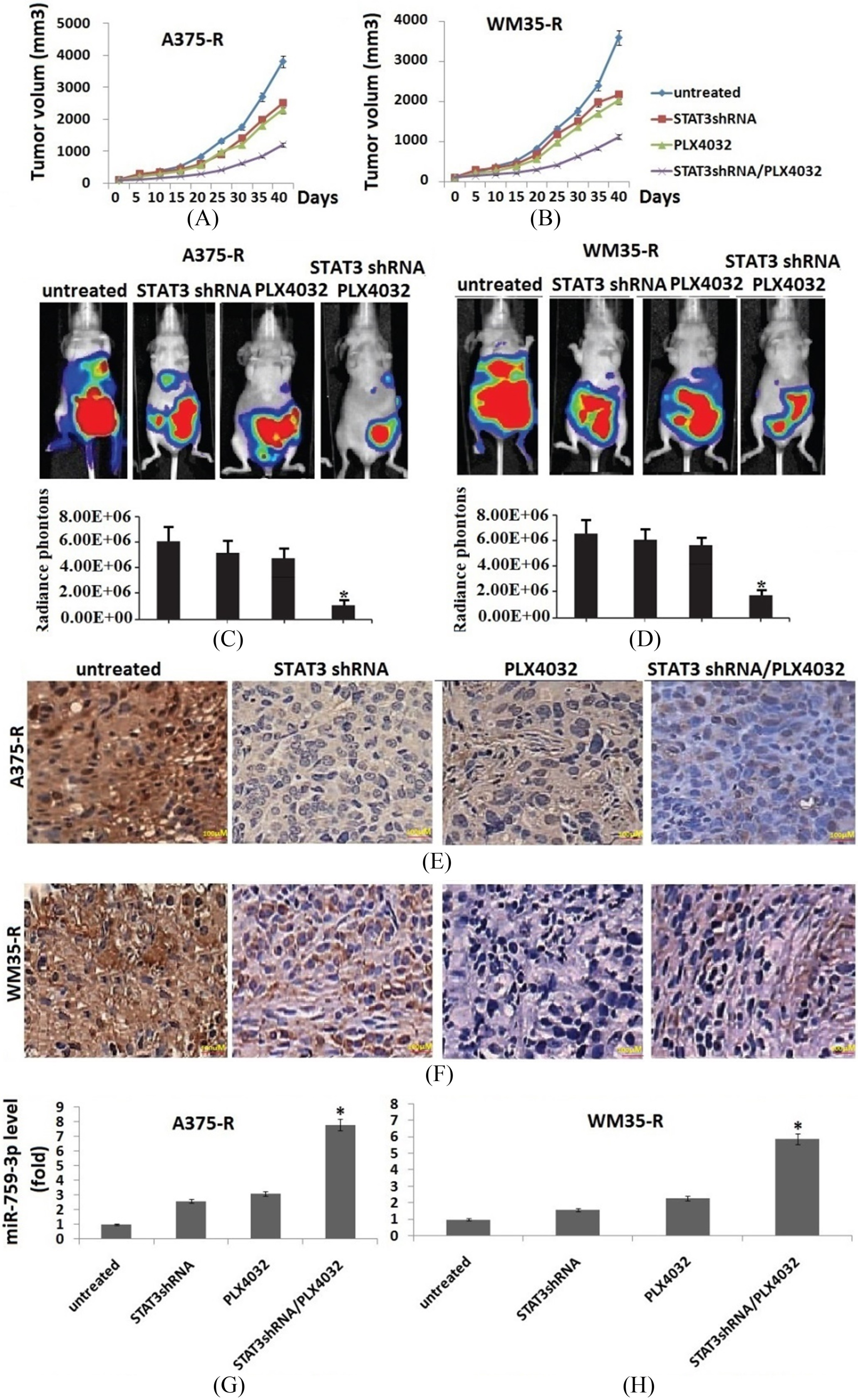
continued, I, Cell apoptosis was detected by TUNEL staining in A375-R cells transplanted tumor tissue specimens. J, Cell apoptosis was detected by TUNEL staining in WM35-R cells transplanted tumor tissue specimens. Vs untreated, *: 
To further test if STAT3 pathway mediated BRAF inhibitor resistance, STAT3 was knocked down in resistant cell lines A375-R and WM35-R, and parental A375-S, WM35-S cells (Fig. 2A). The results showed that knockdown of STAT3 in A375-R and WM35-R cells, or A375-S and WM35-S cells significantly sensitized the cells to PLX4032 by cell viability (Fig. 2B and C) and apoptosis assay (Fig. 2D and E). Knockdown of STAT3 decreased IC50 values from
Targeting STAT3 regulates sensitivity of melanoma cells to PLX4032 treatment through miR-579-3p
To further analyze how targeting STAT3 sensitized the resistant cells to PLX4032 treatment, we first detected the miR-579-3p expression in A375-S, WM35-S, A375-R, WM35-R and harboring the STAT3-specific shRNA A375-R and WM35-R cells by qRT-PCR. The results showed that miR-579-3p expression was significantly downregulated in A375-R and WM35-R cells compared to the A375-S and WM35-S cells (Fig. 3A). However, in the STAT3-specific shRNA A375-R and WM35-R cells, miR-579-3p expression was significantly upregulated compared to the untreated A375-R and WM35-R cells (Fig. 3B and C).
To further test whether miR-579-3p upregulated by STAT3 sliencing plays a role in PLX4032 resistance, A375-R, WM35-R or harboring the STAT3-specific shRNA A375-R and WM35-R cells were transfected with anti-miR-579-3p expression vector, then treated with PLX4032 and assessed by TUNEL assay. The result showed that transfection of anti-miR-579-3p in A375-R, WM35-R or harboring the STAT3-specific shRNA A375-R and WM35-R cells decreased miR-579-3p expression (Fig. 3B and C) and induced resistance to PLX4032 (Fig. 3D and E).
Targeting STAT3 sensitizes melanoma cells towards vemurafenib treatment in vivo
In the next step, we analyzed whether STAT3 is functionally involved in the resistance mechanisms to BRAF inhibitors in vivo. The A375-R or WM35-R cells and resistant A375-R or WM35-R cells harboring the STAT3-specific shRNA were subcutaneously injected into the mice, until a median tumor volume of approximately 100 mm
To characterize the mechanism by which targeting STAT3 induces BRAF inhibitor resistance to A375-R or WM35-R cells, the effect of STAT3 on miR-579-3p expression was determined by qRT-PCR. The results showed a significant decrease of miR-579-3p level in A375-R or WM35-R cells that did not receive any further therapy, but significantly increase in vemurafenib or shRNA STAT3 groups alone, especially increased in combined groups (Fig. 4G and H). TUNEL assay showed that the number of apoptotic cells significantly increased in vemurafenib or shRNA STAT3 groups alone, especially increased in combined groups (Fig. 4I and J).
Discussion
Acquired resistance to the second generation BRAF inhibitor vemurafenib is limiting the benefits of long term targeted therapy for patients with malignant melanomas that harbor the V600E BRAFmutation [22]. Since many resistance mechanisms have been described, most of them causing a hyperactivation of the MAPK- or PI3K/AKT signaling pathways, one potential strategy to overcome BRAFi resistance in melanoma cells would be to target important common signaling nodes [23, 24]. Known factors that cause secondary resistance include the overexpression of receptor tyrosine kinases (RTKs), alternative splicing of BRAF or the occurrence of novel mutations in MEK1 or NRAS [23].
The STAT3 pathway has been shown to be activated in many types of cancer and is associated with cancer transformation, angiogenesis, invasion, and metastasis and with immune system suppression [25]. Our present results provide additional insight into the complexity of the vemurafenib resistance phenotype of melanomas and advance the basis upon which such resistance may be overcome. The first major finding in this work is that STAT3 was activated in the vemurafenib-resistant melanomas cells in vitro and in vivo. We clearly show that reduced pSTAT3 levels by targeting STAT3 can strongly enhance the growth inhibitory and apoptotic effects of vemurafenib in these vemurafenib-resistant or sensitive cells. This suggests that pSTAT3 plays a role in a subset of melanoma cells with acquired resistance towards vemurafenib, and knocking down of STAT3 inhibits the development of resistance to vemurafenib.
Although targeting STAT3 combined with the BRAF V600E inhibitor vemurafenib provides therapeutic benefits but the underlying mechanisms remains unknown. Small noncoding microRNAs (miRNAs) have been confirmed to regulate the expression of target mRNAs by repressing their translation [26]. A growing body of evidence shows that dysregulation of miRNA expression contributes to acquisition of drug resistance by cancer cells [27]. Nevertheless, relatively few studies have explored the roles of miRNAs in resistance to BRAF inhibitor therapy, although several studies identified miRNAs that alter some of the oncogenic factors in melanoma cells [28].
miR-579-3p has been found to act not only as an oncosuppressor whose downregulation is linked to the progression of metastatic melanoma, but also as a factor contributing to the development of drug resistance, and its overexpression was able to reduce melanoma cell growth both individually and in combination with BRAF inhibitor vemurafenib [29]. In the present study, we found that targeting STAT3 by shRNA alone or vemurafenib alone induced miR-759 expression in vemurafenib-resistant melanomas cells in vitro and in vivo. Combined treatment with STAT3 shRNA and vemurafenib further increased the expression level of miR-759. However, targeting miR-579-3p expression was able to revert drug resistance to a BRAF inhibitor in combination with STAT3 sliencing and, more importantly, to reverse the establishment of acquired drug resistance, suggesting that STAT3 regulates miR-579-3p, which affected the sensitivity of melanoma cells to BRAF inhibitor vemurafenib. In this regard it will be important in the future to investigate in detail the mechanisms responsible for miR-579-3p expression and how these are affected during melanoma progression and development of drug resistance. miR-579-3p could be regulated by specific transcription factors, whose expression is altered during the development of drug resistance.
In summary, we found that the activation of STAT3 pathways mediates vemurafenib resistance. We further found that STAT3-mediated vemurafenib resistance occurs through the inhibition of miR-579-3p. Our results suggest that the combination of STAT3 sliencing or miR-579-3p overexpression may be productive approaches for melanoma therapy.
Footnotes
Acknowledgments
This research was supported by Qingdao Basic Science Research Program (No. 15-1-4-161-jch); Shandong Natural Scientific Research Program (No. R2013HQ006).
Conflict of interest
None.