Peroxisome proliferator-activated receptor- (PPAR-) activation has been reported to reduce myocardial ischemia-reperfusion (I/R) injury by inhibiting cell apoptosis. However, the antiapoptotic mechanism of PPAR- is still unknown. Fenofibrate is a PPAR- agonist In the present study, we investigate the effects and relevant mechanism of fenofibrate on experimental myocardial ischemia-reperfusion (I/R) injury in rats.
METHODS:
Adult male Wistar rats were pretreated with fenofibrate (80 mg/kg) daily for a period of 7 days. After the treatment period, myocardial I/R injury model was made by left anterior descending coronary artery ligation for 45 min and reperfusion for 120 min. Myocardial infarct size, malondialdehyde (MDA) cleaved-caspase-9 protein expression, PPAR and uncoupling protein 2 (UCP2) mRNA levels in myocardial tissue were detected Cell apoptosis was detected by Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL). Serum lactate dehydrogenase and creatine kinase activities were measured in rats pretreated with fenofibrate The ultrastructure of myocardial tissues was observed.
RESULTS:
Significant increases in myocardial cell apoptosis, malondialdehyde (MDA) level and cleaved-caspase-9 protein expression level in myocardial tissue were observed, along with reductions of PPAR and uncoupling protein 2 (UCP2) mRNA levels in myocardial tissue of the experimental myocardial ischemia-reperfusion (I/R) injury in rats. Impaired mitochondria were also observed under electron microscopic. However, pretreatment of ischemia/reperfusion rats with fenofibrate brought the biochemical parameters and related genes expression levels to near normalcy, indicating the protective effect of fenofibrate against myocardial ischemia/reperfusion injury in rats.
CONCLUSIONS:
The PPAR- activator fenofibrate conferred cytoprotective effect against myocardial ischemia-reperfusion (I/R) injury in rats. Associated mechanisms involved decreased cleaved-caspase-9 expression and decreased cell apoptosis.
Myocardial ischemia/reperfusion (I/R) injury contributes to adverse cardiovascular outcomes after myocardial ischemia, cardiac surgery or circulatory arrest [1]. Despite years of intensive investigation, we are still far away from thoroughly understanding the underlying mechanisms of I/R.
Ischemia and reperfusion activates various programs of cell death, which can be categorized as necrosis, apoptosis or autophagy-associated cell death [2]. Recent studies have shown that inhibition of apoptosis may have promise as a therapeutic strategy for ischemia-reperfusion injury. For example, a study in a mouse model of acute kidney injury identified the matricellullar protein thrombospondin 1 (THBS1, also known as TSP-1), produced by injured proximal tubular cells, as an inducer of apoptosis and found that Thbs1 mice are protected from injury [3]. Other studies have focused on platelet-derived growth factor CC (PDGF-CC), a potent neuro-protective factor that acts by modulating glycogen synthase kinase 3 (GSK-3) activity [4]. PDGF-CC gene or protein delivery protected neurons from apoptosis in both the retina and brain in various animal models of neuronal injury, including ischemia-induced stroke. PDGF-CC treatment resulted in increased levels of GSK-3 Ser9 phosphorylation and decreased levels of Tyr216 phosphorylation, consistent with previous findings that Ser9 phosphorylation inhibits and Tyr216 phosphorylation promotes apoptosis [4, 5]. We therefore suggested that inhibition of myocardial cell apoptosis could also alleviate myocardial I/R injury.
Peroxisome proliferator-activated receptor (PPAR) is a ligand-activated transcription factor, belonging to the nuclear receptor superfamily. Millions of patients are treated with fibrate PPAR- activators for dyslipidemia, and many of these patients have, or are at risk to develop, ischemic heart disease. Although there is prominent expression of PPAR- in myocardium [6], the function of PPAR- in the heart remains uncertain. In particular, it is unknown whether activation of PPAR- modifies responses to myocardial ischemia-reperfusion (I/R) injury.
Fenofibrate, a peroxisome proliferator-activated receptor alpha (PPAR-) agonist, had been approved by the Food and Drug Administration for clinical use in patients with hypertriglyceridaemia and mixed dyslipidaemia for decades. Besides, fenofibrate was renal protective in doxorubicin-induced glomerular injury [7] and cisplatin-induced proximal tubule cell death [8]. Several studies in rodents have indicated that pharmacological activation of PPAR- by Fenofibrate is protective in myocardial I/R, helping to preserve contractile function and/or reduce infarct size [9, 10]. On the other hand, transgenic mice with cardiac-specific overexpression of PPAR- develop a cardiomyopathy characterized by excessive myocardial neutral lipid accumulation, and PPAR- null mice have better functional recovery from I/R than wild-type mice [11, 12]
In the present study, we investigate the effects of PPAR- activation by Fenofibrate in myocardial I/R injury in rats evaluating the potential mechanism involved.
Materials and methods
Animal
Male Wistar rats (160–220 g body weight), fed a standard diet and tap water ad libitum. All animals were kept under standard conditions with a constant 12 (light): 12 h (dark) cycle (lights on at 06:00 h) and temperature (22C 2C) were employed. All of the animal procedures were approved by an institutional animal care and use committee and were performed according to the Guideline of the Ethical Committee of Harbin Medical University.
Myocardial I/R injury model
Wistar rats were randomly divided into four groups ( 8): 1) normal group (Normal); 2) sham group (Sham); 3) I/R group (I/R); 4) fenofibrate pre-treatment I/R group (FF I/R). Fenofibrate (Sigma, USA) was suspended in 3% Gum acacia and administered for 7 days at the dose of 80 mgkgday by gavage. In the rest three groups, rats were given a similar amount of the solvent (3% Gum acacia). The dose of fenofibrate and the concentration of Gum acacia were based on our previous study [12]. One hour following the last intragastric administration, myocardial I/R model was performed. Briefly, rats were anesthetized by intraperitoneal injection of 10% chloral hydrate (3 ml/Kg). After the oral endotracheal intubation, the animals were mechanically ventilated with room air by a rodent ventilator (room air, rate 75 cycles/min, 3 ml/100 g tidal volume). ECG was recorded. Body temperature was maintained at 37C 0.5C by a heated operation table. The heart was exposed through a left thoracotomy and the left anterior descending (LAD) coronary artery was ligated (1–2 mm region under the boundary pulmonary artery pyramid and left auricle of heart) with a 5-0 polyester suture. A small polyethylene tube was placed between the ligature and myocardial tissues. The rats in sham group underwent the same surgical procedures, except the suture was not fastened. After 45 min ischemia, the ligature was released to permit reperfusion for 120 min. The reperfusion was associated with hyperemia and the disappearance of the cyanotic color of the myocardium. After 45 min of acute myocardial ischemia and 120 min reperfusion, 5 mL of blood was taken from abdominal aorta, standing at 4C for 1 hour, then centrifuged at 10,000 rpm for 5 minutes. Serum was frozen at 20C until LDH and CK activity were assessed. The heart was excised and the ischemic area of the left ventricle was removed and frozen in liquid nitrogen immediately or myocardial homogenate for further measurement.
Serum myocardial enzyme activity and the MDA level in myocardial tissue determination
The serum LDH and CK activity were measured by LDH assay kit (Jiancheng, China) and CK assay kit (Jiancheng, China).The level of Malondialdehyde (MDA) in the myocardium was measured using a MDA assay kit (Beyotime Biotechnology, China), according to the manufacturer’s protocol. Heart tissues were homogenated with lysis buffer and centrifuged at 1600 g for 10 minutes at 4C to get supernatant. The protein concentration of the supernatant was measured by Bicinchoninic acid kit (Beyotime biotechnology, China). Then the thiobarbituric acid (TBA) was put into the supematant, and the reaction products were measured with Varioskan Flash microplate reader at 532 nm emission wavelength and 450 nm emission as reference wavelength. The MDA level was expressed as ng/mg protein.
2,3,5-triphenyltetrazolium chloride and Evans blue staining
The infarct size (IS) and area at risk (AAR) size were evaluated by double staining with 2,3,5-tripheny-ltetrazolium chloride (TTC)/Evans blue and determined by a computerized planimetric method, as described earlier [13]. After 120 min of reperfusion, the LAD was re-ligated, and 1 ml of 5% Evans blue was injected via the left femoral vein to delineate the ischemic myocardium (area at risk, AAR). The rats were euthanized with potassium chloride injection, the heart was rapidly excised and placed in 20C for 10 min. The heart was cut into1-2 mm-thick slices perpendicular to the heart base-apex axis. The slice were photographed and then put into 1% 2,3,5-triphenyltetrazo-lium chloride (TTC) for 15 min at 37C to identify the infarction area (INF). After an overnight incubation in 4% formaldehyde, the slices were photographed again. AAR, INF and the left ventricular area (LV) were determined by planimetry of computer images (Image pro-plus). The IS was expressed as the ratio of INF/AAR.
Myocardial apoptosis determination
The cardiomyocyte apoptosis was determined by using a cell death detection kit (Roche, USA). After 120 min reperfusion, heart tissue distal 2–3 mm to the ligature of left ventricular anterior wall was cut and trimmed into 5 mm and fixed in 4% (vol/vol) paraformaldehyde for 24 hours, then paraffin embedded. Sections of 5-m thickness were mounted on gelatin-coated glass slides. And the serial sections were performed according to the manufacturer’s recommendations. The terminal deoxynucleotidyl trans-ferase-mediated dUTP nick-end labeling (TUNEL) positive cell nuclei present as brown, the normal nuclei was blue. Five fields were selected from every slice, and the number of TUNEL-positive nuclei was calculated by Image Pro Plus software.
Electron microscopy
At the end of reperfusion, the tissue of cardiac apex was excised and trimmed into 1 mm and fixed overnight in 3% glutaraldehyde at 4C. Trimmed tissues were post-fixed by 1% osmium tetraoxide for 2 h and prepared for thin sectioning. The thin sections were observed with a transmission electron microscope.
Immumohistochemical analysis
Tissues of 4 m were cut and transferred to 3-amino-propyl-triethoxy-silane coated slides and incubated overnight at room temperature. Antigen retrieval of sections immersed in citrate buffer solution was done using a pressure cooker. Endogenous peroxidases were blocked (Novacastra, Leica Systems, UK) at room temperature for 15 min. Then sections were incubated with primary anti-cleaved-caspase-9 (Ruiying, China, 1:200) for 1 h followed by incubation with HRP)-conjugated goat anti rabbit IgG (ZSGB BIO, China) for 30 min. Then a drop of streptavidin was added from secondary antibody kit) for 30 min followed by incubation with 3’diaminobenzidine-tetrahydrochloride for 5–10 min. Then the sections were counterstained with hematoxylin and mounted.
The effect of pretreated with fenofibrate on myocardial infract size of rats with acute I/R. A. Representative slice of myocardium stained by TTC/Evans blue. The region with blue indicates the non-ischemic area, the red indicates the area at risk, and the white indicates the infracted area. Bar 5 mm. B. The quantitative analysis of area at risk, expressed as the percent of left ventricle. C. The quantitative analysis of infarct size, expressed as the percent of the area at risk. Data are presented as mean SEM ( 8). Significant differences ***: 0.001 compared with the sham group, : 0.001 compared with the I/R group.
The stained sections were scanned under low power to determine the area that stained brown color and was considered as positive for cleaved-caspase-9 expression. Cytoplasmic and membranous staining were considered as positive immunoreaction for cleaved-caspase-9 In a randomly selected five fields, 100 cells were considered in each field. Out of 100 cells cleaved-caspase-9 positively stained cells were counted. Two observers evaluated all the slides. The cleaved-caspass-9 expression intensity was expressed as Inte-grated Optical density (IOD), as detected using a software for image analysis (Image Pro Plus 6.0).
Reverse transcription polymerase chain reaction (RT-PCR) and real-time quantitative PCR
Heart tissue were collected and frozen at 80C for evaluating the expression of PPAR and UCP2 mRNA. The total RNA was extracted using Trizol reagent (Tiangen, China), and the reverse transcription of the total RNA to cDNA was carried out using an AccuPower RocketScriptTM RT PreMix Kit (Bioneer, South Korea) respectively following the instructions of manufacturer. The thermal cycle profile for reverse transcription was set for primer annealing at 37C for 10 min, cDNA synthesis at 50C for 60 min, heat inactivation at 95C for 5 min. The real-time quantitive PCR was performed using AccuPower Plus DualStarTM qPCR PreMix Kit (Bioneer, South Korea) in a Bio-Rad iQ5 optical module. The cycling conditions were set for pre-denaturation at 95C for 10 min; 40 cycles of denaturation at 95C for 10 sec; annealing at 58C for 20 sec. The primer sequences are as follows: primer of PPAR forward: 5’-TGGCGTTCGCA-GCTGTTTTG-3’ and reverse 5’-CTCGTGTGCCC-TCCCTCAAG-3’; primer of UCP2 forward: 5’-GC-AGTTCTACACCAAGGGCT-3’ and reverse 5’-GG-AAGCGGACCTTTACCACA-3’; pr- imer of -actin-forward: 5’-CACCCGCGAGTACAA CCTTC-3’ and reverse 5’-CCCATACCCACCATCAC ACC-3’.
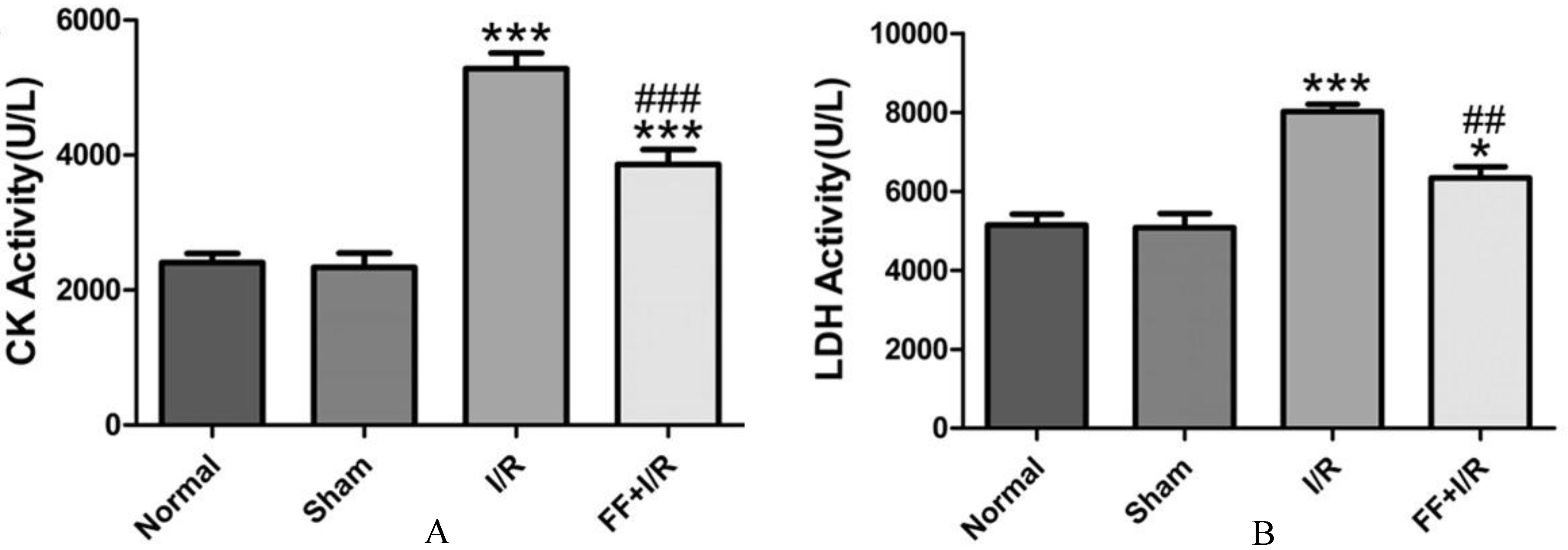
The effect of pretreated with fenofibrate on the activities of serum myocardial enzyme of rats with acute I/R. A. The serum CK activity. B. The serum LDH activity. Data are presented as mean SEM ( 8). Significant differences, ***: 0.001, compared with the sham group, *: 0.05,compared with the sham group, ###: 0.001, compared with the I/R group, ##: 0.01,compared with the I/R group.
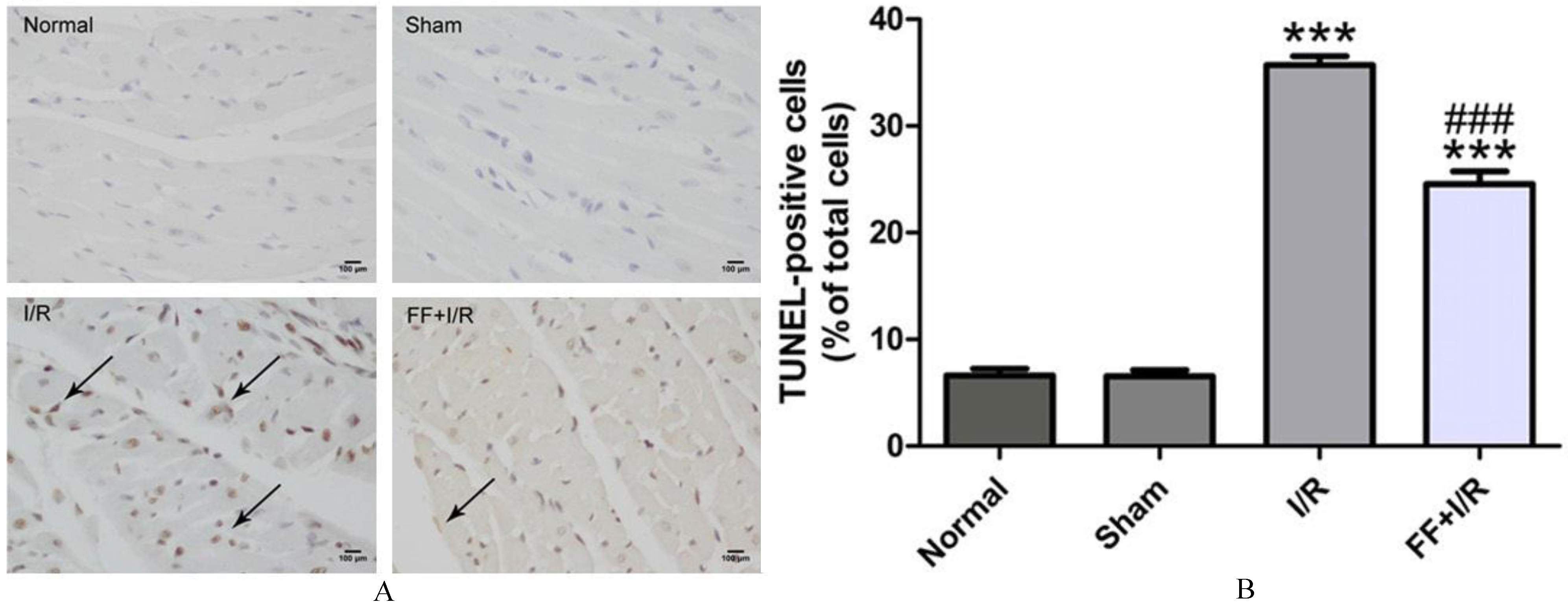
The effects of pretreated with fenofibrate on cardiomyocyte apoptosis of rats with acute I/R. A. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)-stained myocardium of rats from each group. Arrows indicate TUNEL-positive cells. Bar 100 m. B. The quantitative analysis of TUNEL-positive cells. Data are presented as mean SEM ( 8). Significant differences. ***: 0.001 compared with the sham group, ###: 0.001 compared with the I/R group.
Statistical analysis
All data were expressed as mean SEM. A Student’s t-test or one-way ANOVA on ranks was performed to compares the statistical difference between the groups with Graphpad Prim 5. 0.05 was considered statistically significant.
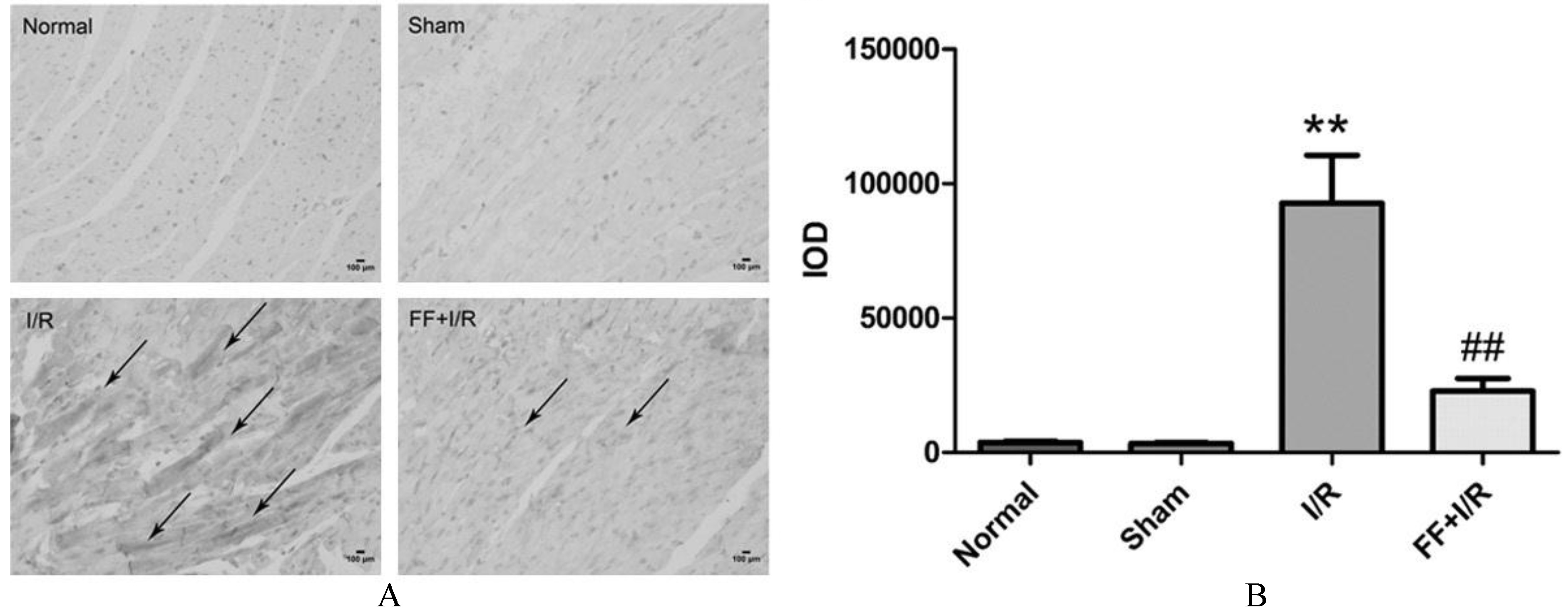
The influence of pretreated with fenofibrate on the expression of cleaved-caspase-9 in myocardium of rats with acute I/R. A. Representative photomicrographs of immunohistochemistry staining for cleaved-caspase-9. Bar 100 m. Arrow indicates cleaved-caspase-9 positive staining. B. Quantitative analysis of cleaved-caspase-9 positive staining in each groups. Data are presented as mean SEM ( 8). Significant differences, **: 0.01 compared with the sham group; ##: 0.01, compared with the I/R group.
Results
PPAR activation protected against acute myocardial I/R injury
The infarct size and activity of serum myocardial enzyme were performed to assess acute myocardial I/R injury. Both the area at risk and infarct size were shown in Fig. 1A. There was no difference in ratio of AAR to LV between the I/R group and the fenofibrate pretreatment group (I/R: 47.84 2.94% vs. FF I/R: 49.15 2.32%, 0.05; Fig. 1B), indicating a similar tension and placement of the ligature among the groups. But fenofibrate pretreatment led to significant reduction in infarct size (IS/AAR, FF I/R: 32.27% 2.11% vs. I/R: 47.09 3.34%; 0.05; Fig. 1C) in comparison with the I/R group. Compared to the I/R group, the serum CK activity (I/R: 8018.82 183.42 vs. I/R FF: 6336.12 690.96, 0.05; Fig. 2A) and LDH activity (I/R: 8018.82 183.42 vs. I/R FF: 6336.12 690.96, 0.05; Fig. 2B) were significantly decreased in fenofibrate pretreatment group, suggesting that PPAR activation protects myocardium from acute I/R injury.
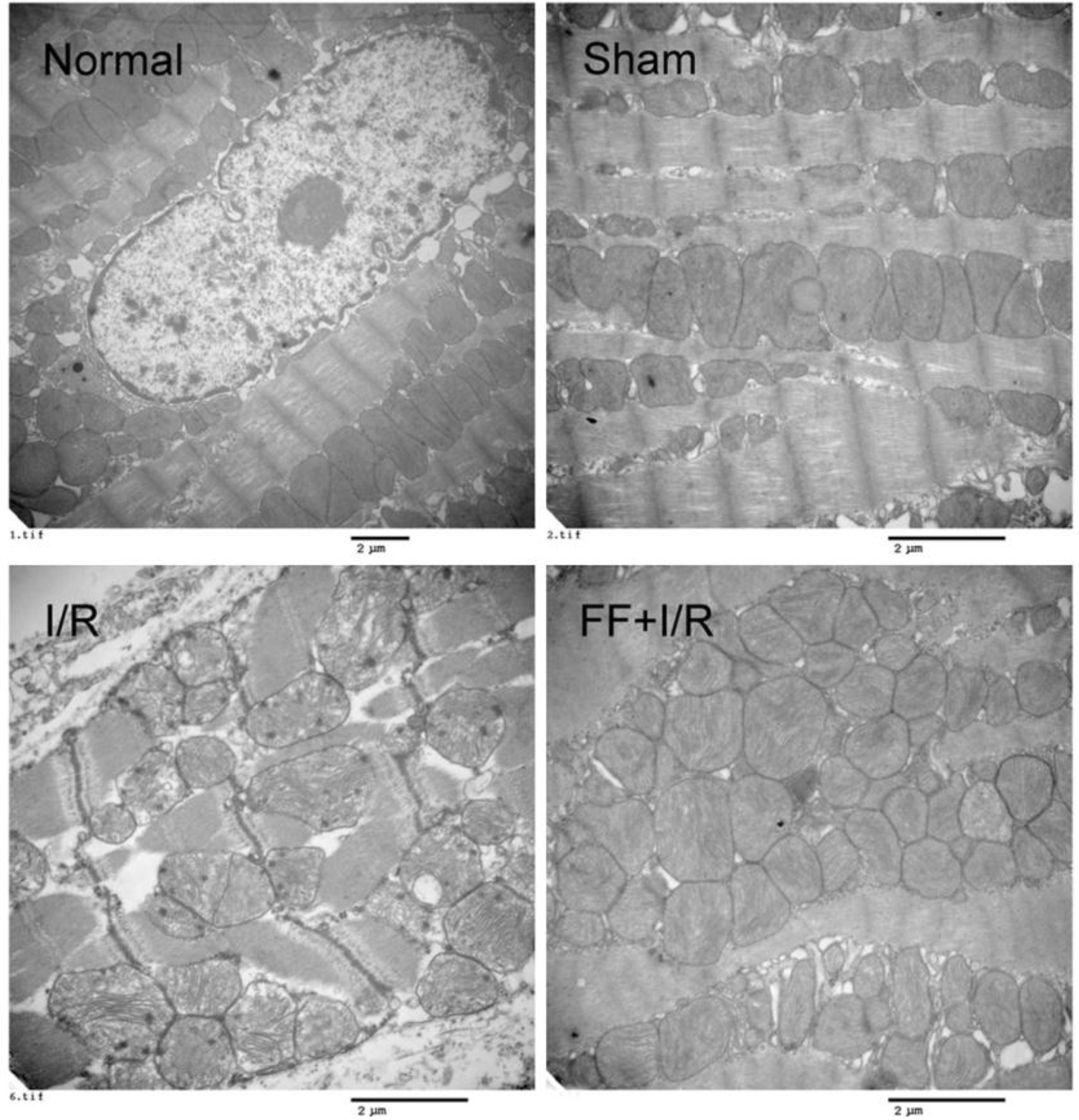
The influence of pretreated with fenofibrate on the ultrastructure of myocardial tissues of rats with acute I/R. The representative electron micrographs of cardiomyocytes of rats in each group. Bar 2 m.
TUNEL staining and caspase-9 activity were performed to measure the extent of apoptosis in ischemic myocardium. As shown in Fig. 3, compared to the I/R group, fenofibrate pretreatment significantly reduced TUNNEL-po-sitive cardiomyocytes ( 0.05). Furthermore, the casepase-9 activity, represented as the cleaved-caspase-9 expression in myocardium, was obviously reduced in fenofibrate pretreated group compared to I/R group ( 0.01, Fig. 4). These results suggests that the participation of anti-apoptosis effect of PPAR in protecting against acute myocardial I/R injury.
Mitochondrial ultrastructure were observed by transmission electron microscopy (Fig. 5). Results showed that I/R resulted in distinctive ultrastructure alterations, including disordered mitochondrion distribution with disarranged and obscure crista and vacuoles within the matrix, accompanying by disrupted myofilament and sarcomere. Noticeably, fenefibrate pretreated alleviated these deleterious effects of mitochondrial ultrastructure induced by I/R injury.
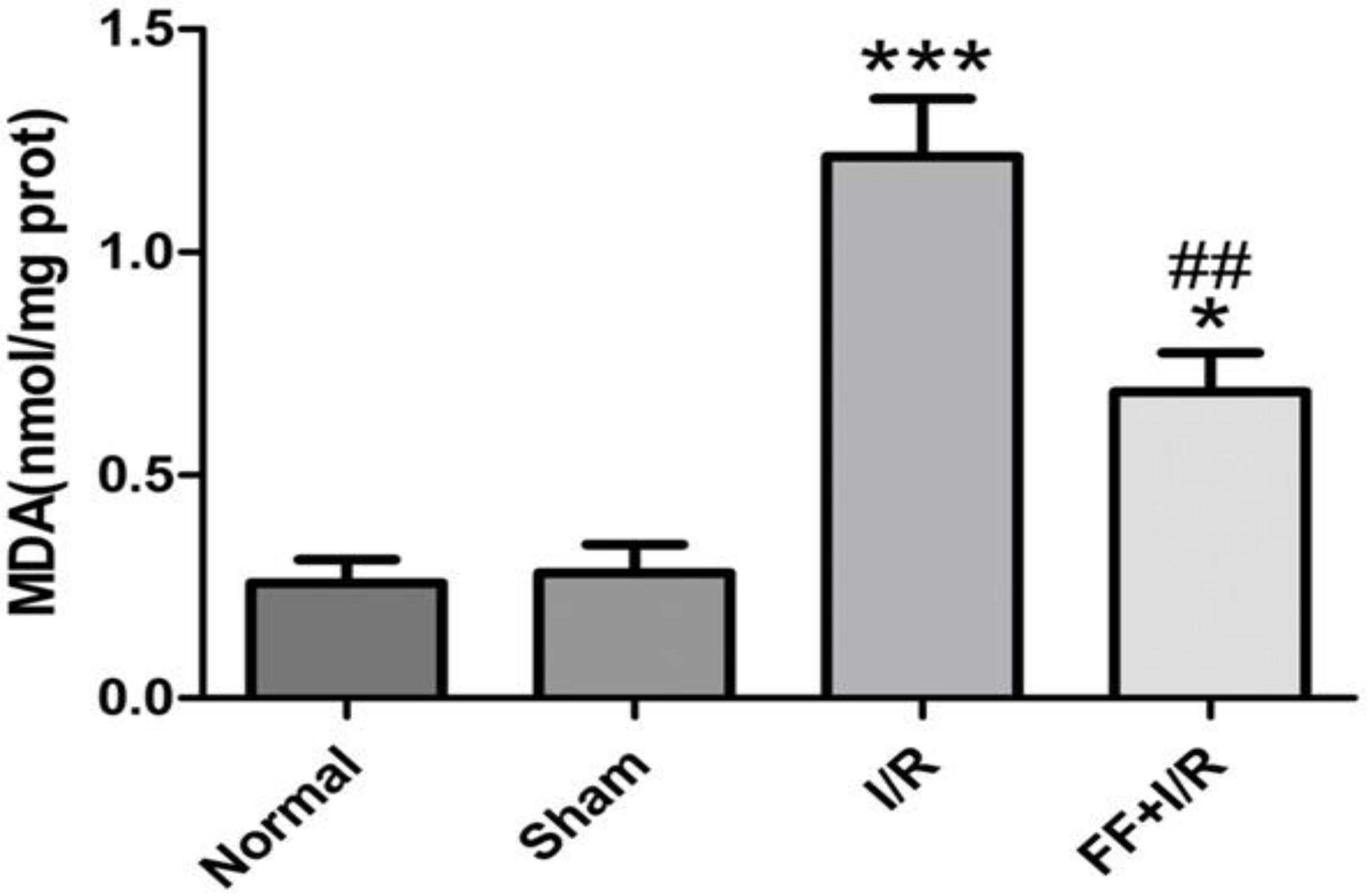
We measured the malondialdehyde (MDA) level in ischemic myocardium, and we found that the production of MDA significantly increased in I/R-treatment group, compared with sham group (Sham: 0.28 0.06 vs. I/R: 1.21 0.13, 0.001). While pretreatment with fenofibrate significantly reduced the elevated MDA production induced by I/R (I/R: 1.21 0.13 vs. I/R FF: 0.69 0.09, 0.01, Fig. 6).
The effect of fenofibrate pretreated on MDA level in the myocardium of rats with acute I/R. Data are presented as mean SEM ( 8). Significant differences, ***: 0.001 vs. Sham group. *: 0.05 vs. Sham group. ##: 0.01 vs. I/R group.
The mRNA expression of PPAR and UCP2 in myocardium
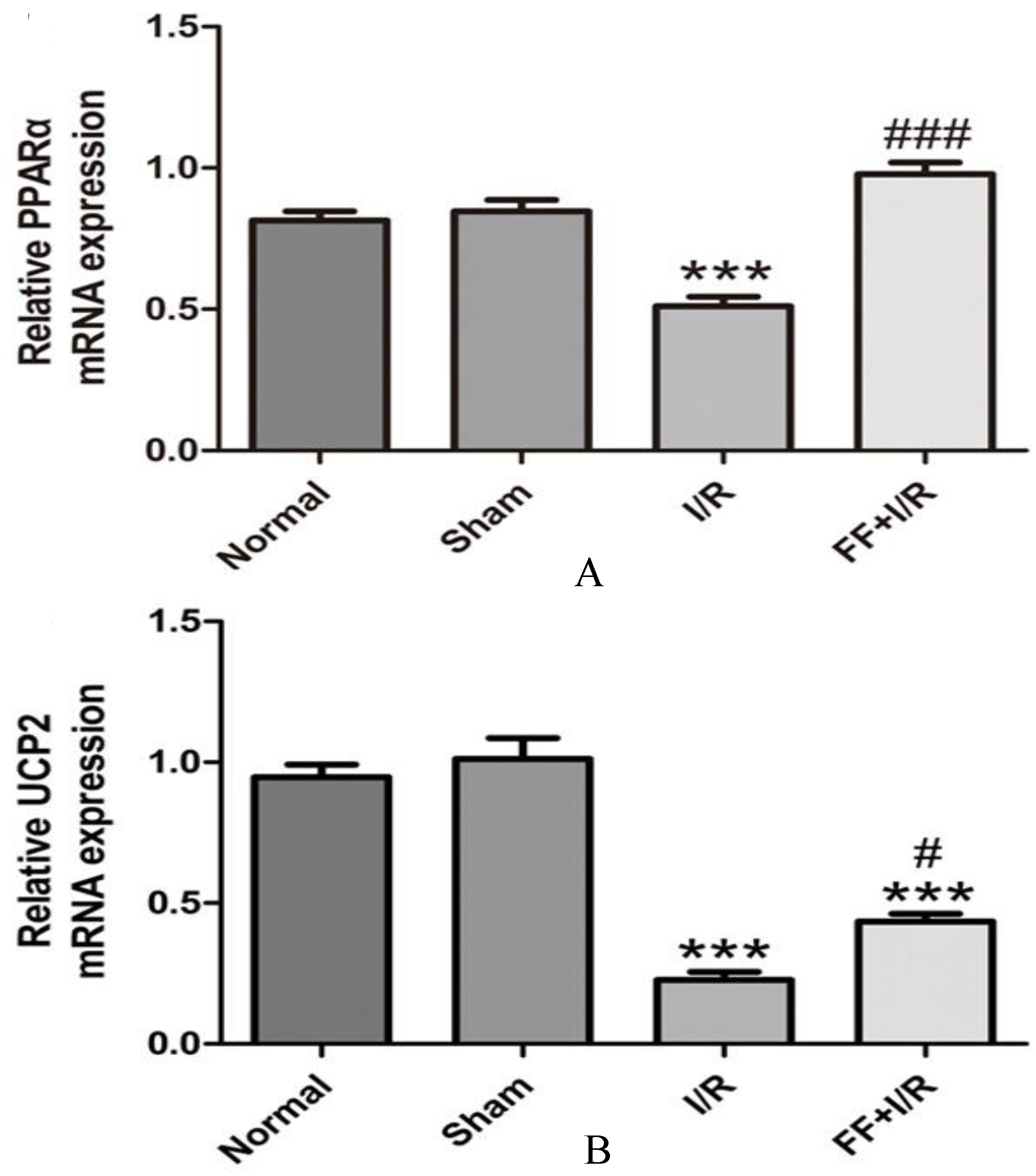
As shown in Fig. 7, compared with the normal group, the expression of PPAR mRNA (Normal: 0.79 0.04 vs. Sham: 0.85 0.04, 0.05) and UCP2 mRNA (Normal: 0.94 0.05 vs. Sham: 1.01 0.07, 0.05) in myocardium has no significantly difference in the sham group. Compared with the sham group, the expression of PPAR and UCP2 mRNA significantly decreased ( 0.001, 0.001) in the I/R group. Compared with the I/R group, pretreated with fenofibrate, significantly upregulated the expression of PPAR mRNA (I/R: 0.51 0.02 vs. I/R FF: 0.98 0.04, 0.001) and UCP2 mRNA (I/R: 0.23 0.03 vs. I/R FF: 0.39 0.03, 0.01).
The effect of fenofibrate pretreated on expression of PPAR and UCP2 mRNA in the myocardial tissue of rats with acute I/R. The quantitative fold changes in mRNA expression were determined relative to -actin mRNA levels in each corresponding group and were calculated using the 2-CT method. A. The expression of PPAR mRNA in myocardial. B. The expression of UCP2 mRNA in myocardial. Data expressed as mean SE. ( 8) ***: 0.001 vs. Sham group. ###: 0.001 vs. I/R group. #: 0.05 vs. I/R groups.
Discussion
We discovered in our previous research that pretreatment with fenofibrate could remarkably improve isoprenaline-induced acute myocardial ischemia injury. We explored the nonmetabolic effect of PPAR on rat with acute myocardial I/R injury and the potential mechanism involved in this paper. Consistent with the previous investigation, the results in this paper demonstrated that the expression of PPAR mRNA decreased in the myocardial tissue of I/R rats. Short-term pretreatment with fenofibrate, a specific ligand of PPAR to activate PPAR, notably reduced the myocardial infarction size, decreased the activity of serum myocardial enzyme, and reduced the MDA level in myocardium, alleviated the cardiomyocyte mitochondria damage, decreased myocardial cell apoptosis, and displayed protective effect on acute myocardial I/R injury. At the early stage of reperfusion, ROS is massively produced and accumulated, inducing polyunsaturated fatty acid oxidation and damaging the cell membrane directly through lipid peroxidation [14], as well as producing toxic metabolites, such as MDA [15]. MDA is a marker reflecting the degree of lipid peroxidation injury [16]. We found that the content of MDA was distinctly increased in ischemia reperfusion myocardial tissue, indicating remarkable oxidative stress injury, while activation of PPAR could reduce the MDA content in myocardial tissue from ischemia reperfusion rats, indicating that PPAR activation could alleviate ischemia-reperfusion induced oxidative stress injury.
The mechanism of anti-oxidant activity of PPAR in heart remains unclear. Previous studies mostly focus on regulating the expression and activity of antioxidant enzymes, such as SOD and catalase (CAT) [17, 18]. PPAR activation in liver upregulates the expression of uncoupling protein 2 (UCP2), and relieves the oxidative stress induced injury [19]. UCP2 is one of the members of anion carrier protein family, and locates in the inner membrane of mitochondrial. By regulating the transfer of proton to the mitochondrial matrix, UCP2 dissipates the proton gradient in the inner mitochondrial membrane, decreases the mitochondrial inner membrane potential, and consequently reduces the production of ROS and alleviates oxidative stress response [20, 21]. The generation of mitochondrial ROS during I/R is related to a specific metabolic process [22]. The succinate that is massively produced and accumulated at stage of myocardial ischemia, is rapidly oxidized after reperfusion, which drives reverse electro transport at complex I, thus increasing the mitochondrial inner membrane potential and inducing the production of mitochondrial ROS that initiates I/R injury. UCP2 can mediate proton leak to dissipate the mitochondrial proton-motive force, so we detected the expression of UCP2 in myocardial tissue. We found that the mRNA level of UCP2 was decreased in the myocardial tissue suffered from I/R, while PPAR activation could apparently upregulate the UCP2, indicating that UCP2 may participate in the antioxidant effect of PPAR on acute myocardial I/R injury. In addition, we found that the expression of PPAR mRNA was significantly increased in the I/R groups, however, PPAR mRNA was significantly decreased in the fenofibrate pre-treated groups compared to the I/R groups. PPAR mRNA in myocardium has no difference between the sham group and the fenofibrate pre-treated group, but the mRNA expression of UCP2 failed to keep parallel to the expression of PPAR, indicating that there were other factors involved in regulating of UCP2. The role of UCP2 in myocardial I/R injury remains further investigation.
Apoptosis is the characteristic change of I/R injury, and inhibition of myocardial apoptosis can alleviate myocardial I/R injury [23]. The intrinsic apoptosis is a mitochondrion-centered cell death that is mediated by mitochondrial outer membrane permeabilization (MOMP), results in apoptosome formation, activation of caspase-9 [24, 25] Once caspase-9 was active, caspase-9 can directly cleave and activate caspase-3 and caspase-7. In addition, caspase-9 can prevent accessibility of cytochrome to complex III in the mitochondria, resulting in increased ROS production, but in the presence of effector caspase activity, ROS production is terminated [26].
In this paper, we observed that PPAR activation could alleviate the acute I/R induced injury of mitochondrial ultrastructure, decrease the casepase-9 activity in myocardial tissue, and inhibit myocardial apoptosis. Consequently, PPAR activation might reduce the I/R induced myocardial infarction area by decreasing myocardial apoptosis, and such anti-apoptosis protective effect might be related to alleviating mitochondrial injury induced by oxidative stress.
To sum up, this research sheds new light on the protection effects of PPAR activation on acute myocardial I/R injury, which may be achieved through alleviating oxidative stress induced mitochondrial apoptotic pathway; in addition, such protective effect may be related to the up-regulated expression of UCP2.
References
1.
HausenloyD.J. and YellonD.M., Myocardial ischemia-reperfusion injury: a neglected therapeutic target, J Clin Invest123(1) (2013), 92–100.
2.
HotchkissR.S.StrasserA.McDunnJ.E. and SwansonP.E., Cell death, N Engl J Med361(16) (2009),1570–1583.
3.
ThakarC.V.ZahediK. and ReveloM.P., Identification of thrombospondin 1 (TSP-1) as a novel mediator of cell injury in kidney ischemia, J Clin Invest115(12) (2005), 3451–3459.
4.
TangZ.ArjunanP. and LeeC., Survival effect of PDGF-CC rescues neurons from apoptosis in both brain and retina by regulating GSK3beta phosphorylation, J Exp Med207(4) (2010), 867–880.
5.
LiangM.H. and ChuangD.M., Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival, J Biol Chem282(6) (2007), 3904–3917.
6.
FinckB.N. and KellyD.P., Peroxisome proliferator-activated receptor alpha (PPAR alpha) signaling in the gene regulatory control of energy metabolism in the normal and diseased heart, J Mol Cell Cardiol34 (2002), 1249–1257.
7.
ZhouY.KongX.ZhaoP.YangH.ChenL. and MiaoJ., Peroxisome proliferator-activated receptor-α is renoprotective in doxorubicin-induced glomerular injury, Kidney Int79(12) (2011), 1302–1311.
TaberneroA.SchoonjansK.JeselL.CarpuscaI.AuwerxJ. and AndriantsitohainaR., Activation of the peroxisome proliferator-activated receptor alpha protects against myocardial ischaemic injury and improves endothelial vasodilatation, BMC Pharmacol2 (2002), 10.
10.
YueT.L.BaoW.JuckerB.M.GuJ.L.RomanicA.M. and BrownP.J., Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury, Circulation108(19) (2003), 2393–2399.
11.
FinckB.N.LehmanJ.J.LeoneT.C.WelchM.J.BennettM.J. and KovacsA., The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus, J Clin Invest109(1) (2002), 121–130.
12.
FinckB.N.HanX.CourtoisM.AimondF. and KovacsA., A critical role for PPAR alpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content, Proc Natl Acad Sci U S A100(4) (2003), 1226–1231.
13.
PeiH.YuQ.XueQ.GuoY.SunL. and HongZ., Notch1 cardioprotection in myocardial ischemia/reperfusion involves reduction of oxidative/nitrative stress, Basic Res Cardiol108(5) (2013), 373.
14.
ToyokuniS., Reactive oxygen species-induced molecular damage and its application in pathology, Pathol Int49(2) (1999), 91–102.
15.
ValenzuelaA., The biological significance of malondialdehyde determination in the assessment of tissue oxidative stress, Life Sci48(4) (1991), 301–309.
16.
MengC.LiuJ.L. and DuA.L., Cardioprotective effect of resveratrol on atherogenic diet-fed rats, Int J Clin Exp Pathol7(11) (2014), 7899–7906.
17.
GuellichA.DamyT.ContiM.ClaesV.SamuelJ.L. and PineauT., Tempol prevents cardiac oxidative damage and left ventricular dysfunction in the PPAR-alpha KO mouse, Am J Physiol Heart Circ Physiol304(11) (2013), H1505–H1512.
18.
ZuoN.ZhengX.LiuH. and MaX., Fenofibrate, a PPAR alpha agonist, protect proximal tubular cells from albumin-bound fatty acids induced apoptosis via the activation of NF-kB, Int J Clin Exp Pathol8(9) (2015), 10653–10661.
19.
PattersonA.D.ShahY.M.MatsubaraT.KrauszK.W. and GonzalezF.J., Peroxisome proliferator-activated receptor alpha induction of uncoupling protein 2 protects against acetaminophen-induced liver toxicity, Hepatology56(1) (2012), 281–290.
20.
AzzuV. and BrandM.D., The on-off switches of the mitochondrial uncoupling proteins, Trends Biochem Sci35(5) (2010), 298–307.
21.
MaillouxR.J. and HarperM.E., Uncoupling proteins and the control of mitochondrial reactive oxygen species production, Free Radic Biol Med51(6) (2011), 1106–1115.
22.
ChouchaniE.T.PellV.R.GaudeE.AksentijevićD.SundierS.Y. and RobbE.L., Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS, Nature515(7527) (2014), 431–435.
23.
LiuY.ShiL.LiuC.ZhuG.ZhaoH. and LiS., Effect of combination therapy of propofol and sevoflurane on MAP2K3 level and myocardial apoptosis induced by ischemia-reperfusion in rats, Int J Clin Exp Med8(4) (2015), 6427–6435.
24.
WestermannB., Mitochondrial fusion and fission in cell life and death, Nat Rev Mol Cell Biol11(12) (2010), 872–884.
25.
MiuraT.TannoM. and SatoT., Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis, Cardiovasc Res88(1) (2010), 7–15.
26.
MakazanZ.SainiH.K. and DhallaN.S., Role of oxidative stress in alterations of mitochondrial function in ischemic-reperfused hearts, Am J Physiol Heart Circ Physiol292(4) (2007), H1986–H1994.