
Abstract
BACKGROUND:
Accumulating evidence shows that clinical factors alone are not adequate for predicting the survival of patients with urothelial bladder cancer (UBC), and many genes have been found to be associated with UBC prognosis.
PURPOSE:
The objective of this study is to develop a signature which integrates clinical and molecular information to predict the overall survival of UBC patients more accurate.
MATERIALS AND METHODS:
We integrated messenger RNA (mRNA) and microRNA (miRNA) expression profiles and the corresponding clinical data of 402 UBC patients and 19 normal controls from The Cancer Genome Atlas. Univariate Cox regression followed by a multiple testing correction and an elastic net-regulated Cox regression were adopted to identify a prognostic signature.
RESULTS:
We generated an integrated clinical-RNA signature which consisting of 3 clinical variables, 3 protective mRNAs, 7 risky mRNAs, 2 protective miRNAs and 1 risky miRNA. The area under the receiver operating characteristic curve of the integrated clinical-RNA signature was 0.802, larger than that of the clinical-alone signature (0.709) or the RNA-alone signature (0.726). UBC patients in the high-risk group had a significantly shorter overall survival time compared with patients in the low-risk group (clinical-RNA signature, hazard ratio
CONCLUSIONS:
Our conclusions that we have identified an integrated clinical-RNA signature that was superior to the traditional clinical-alone signature for ascertaining the overall survival prognosis of patients with UBC. These findings provide some novel genes for tumor molecular biologist to further study their functions and mechanisms in UBC tumorigenesis and malignance, and may be useful for effective clinical risk management of UBC patients.
Introduction
Urothelial bladder cancer (UBC) is most common pathological subtype of bladder cancer, and the clinical prognosis of patients with UBC differs significantly which is hard to predict. If a new criterion had been developed to identify the group of high-risk patients ahead, they could get more suitable treatment including extended curative resection or higher radiation/chemo doses, which may greatly ameliorate the poor survival of UBC patients. So, an accurate prediction model is essential for efficient management of UBC which may bring better clinical outcomes to patients with UBC.
A variety of clinical variables in UBC have been studied with regard to their ability to predict disease recurrence, response to therapy, progression, and survival [1, 2, 3, 4, 5]. In the clinical guideline, clinical stage and tumor grade had been recommended as the most important prognostic indicators to forecast disease recurrence and progression [6]. However, significant variability in patient outcome is observed perhaps due to the underlying molecular heterogeneity within the clinically homogeneous tumor groupings.
In recent years, efforts have been made to better understand UBC through basic researches (molecular and genetic), and many studies have revealed that RNAs could serve as novel biomarkers for UBC prognosis [7, 8, 9, 10, 11, 12, 13, 14]. Kim et al. [9] reported that a four-gene signature could predict prognosis of patients with muscle-invasive bladder cancer. Ratert et al. [11] described that expressions of miR-141 and miR-205 were associated with overall survival in patients with bladder cancer. However, these studies mainly focused on messenger RNA (mRNA) profiles or microRNA (miRNA) profiles of UBC genomes independently, and the prognostic signatures were quite different due to a low sample size or the use of an inappropriate regression method for parameter estimation.
Biologically, identification of prognostic signature by integration of mRNA and miRNA expression profiles incorporating more information and may discover more robust prognostic signature due to potential interactions between miRNAs and mRNAs. Methodologically, in genomic expression analysis, there is a so-called “curse of dimensionality” problem in that the number of genes is much larger than the number of samples [15]. In this setting, ordinary regression is subject to over-fitting and instable coefficients, and stepwise variable selection methods do not scale well [16, 17]. Regression by penalization methods has been successfully adapted to high-dimensional multiple genomic datasets and outperforms univariate and multivariate regression methods [18]. At present, the most commonly used penalization methods are ridge regression, Least Absolute Shrinkage and Selection Operator (LASSO) regression and a hybrid of these (elastic net regression); all three methods are based on penalizing the L1 norm, the L2 norm, and both the L1 norm and L2 norm with tuning parameters. Although the traditional Cox proportional hazards model is widely used to discover cancer prognostic factors, it is not appropriate for the genomic setting due to the high dimensionality and colinearity. Several groups have proposed to combine the Cox regression model with the elastic net dimension reduction method to select survival-correlated genes within a high-dimensional expression dataset and have made available the associated computation procedures [19, 20, 21].
By characterizing genetic alterations, epigenetic alterations, and the expression of cancer genomes, The Cancer Genome Atlas (TCGA) project has provided a comprehensive way to understand UBC [22]. As the number of UBC samples in this project grows, the opportunity to identify prognostic molecular signature for patients with UBC is increasing. In the current study, we subjected the integrated mRNA and miRNA profiles of UBC patients to elastic net-regulated Cox regression analysis and identified an integrated prognostic signature that can predict the overall survival (OS) of UBC patients. These may be helpful for selecting high risk UBC patients for better clinical management.
Materials and methods
Patient characteristics and integrated RNA profiles
Clinical data and RNA expression profiles of UBCs were retrieved from TCGA (
Prognostic clinical factors
Survival analysis with log-rank test was preliminarily applied to filter out the demographic and clinical factors that were correlated with the OS of UBC patients from among 9 factors: gender, age at initial diagnosis, clinical stage, tobacco use, anatomic neoplasm subdivision, Karnofsky performance status, human race, lymphovascular invasion, and neoplasm histologic grade. Demographic and clinical factors resulting in a log-rank test P value
Prognostic mRNAs and miRNAs
mRNAs and miRNAs that differentially expressed between UBCs and normal samples were selected using “glmLRT” method implemented in “edger” R package with FDR
Prognosis index and integrated clinical-RNA signature
We adopted a prognosis index (PI) which defined as a linear combination of the observed value weighted by the regression coefficients as indicator for UBC patient prognosis. Specifically,
where, PI
We also used multivariate stepwise Cox regression analysis to determine the prognostic contributions and independence of RNA-PI and the associated clinical factors [15, 23]. RNA-PI and clinical factors with a multivariate Cox regression P value
where, Mean(PI) and sd(PI) are the mean and standard deviation of the PI vector, respectively.
Model evaluation
UBC patients that have WPI
GO and pathway of mRNA and targets of miRNAs
Validated mRNA and targets of miRNAs were available from miRTarBase [25]. Only miRNA and mRNA target interactions with strong evidences, either by reporter assay or western blot, were extracted. Gene ontology enrichment and pathway enrichment were carried out based on these validated targets using online tool STRING [26, 27, 28]. The interactions among products of these targets were visualized by the same tool.
Univariate survival analysis and multivariate Cox regression of demographic and clinical variables
Univariate survival analysis and multivariate Cox regression of demographic and clinical variables
Abbreviation: CI, confidence interval. P-values were obtained from log-like test for survival analysis and Wald test for Cox regression, patients were omitted when data is unavailable. For multivariate Cox regression, variable coding is as follows: Age at diagnosis (1
All statistical analyses were completed by R software [29] (version 3.3.2). Survival analysis with log-rank test was used to compare survival distributions between different groups, and it is implemented by “survfit” function that incorporated in “survival” R package. Univariate Cox regression analysis was used to evaluate the association between RNA expressions across samples with OS of UBC patients. Multivariate Cox regression analysis with step-wise procedure to select independent variables. Both univariate and multivariate Cox regression analyses with Wald test were implemented by “coxph” function that incorporated in “survival” R package. The generalized linear model (GLM) likelihood ratio test is based on the idea of fitting negative binomial GLMs with the Cox-Reid dispersion estimates. The testing can be done by using the “glmLRT” function implemented in “edger” R package [30]. The elastic-net penalized Cox regression was implemented by “glmnet” function and cross validation was implemented by “cv.glmnet” function incorporated in “glmnet” R package. All statistical tests were considered to be significant with
Cross-validation error curve. The left vertical line shows where the cross-validation error curve hits its minimum (lambda 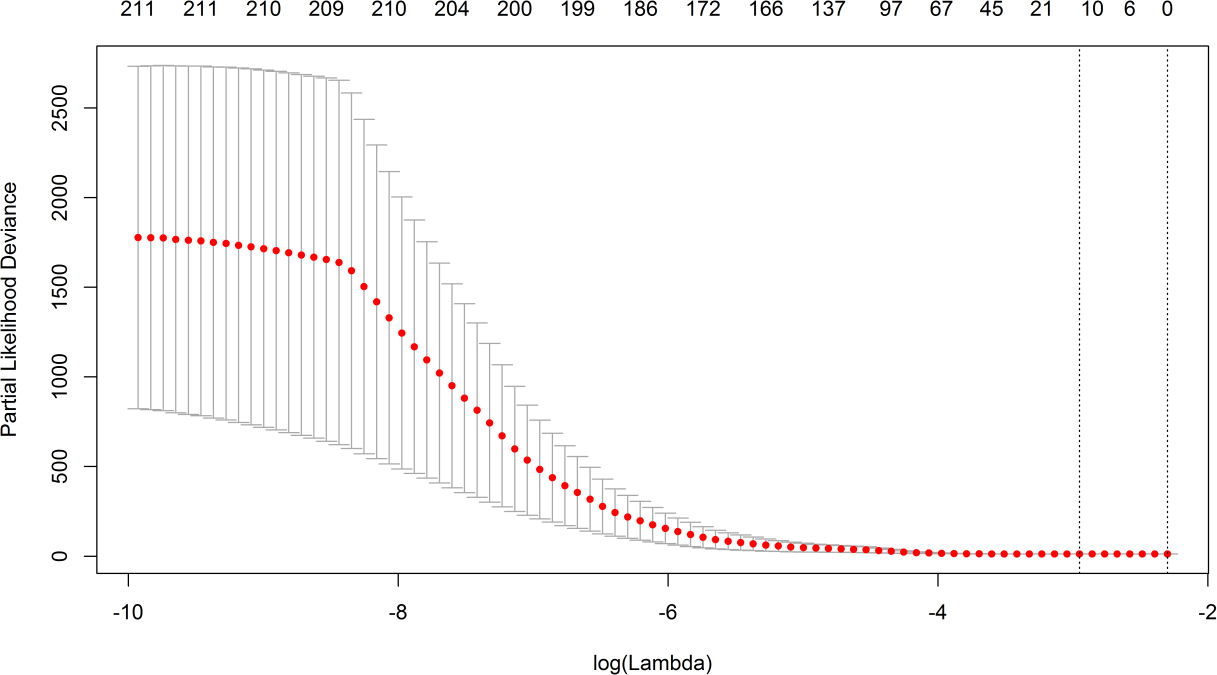
RNAs correlated with UBC prognosis
Clinical characteristics correlated with UBC patient survival
Multivariate Cox regression of demographic and clinical variables
Multivariate Cox regression of demographic and clinical variables
P-values of multivariate Cox regression model were obtained from Wald test.
Of the 402 patients in the TCGA UBC cohort, 176 patients were deceased and 226 were alive at the time of last follow-up. The average age at initial diagnosis of the patients in this cohort was 60.14 years, and the median overall survival time was 813 days (95% confidence interval [CI], 731–895 days). Demographic and clinical data for the TCGA UBC cohort were summarized in Table 1. Gender, age at initial diagnosis, clinical stage, tobacco use, anatomic neoplasm subdivision, Karnofsky performance status, human race, lymphovascular invasion, and neoplasm histologic grade were evaluated by survival analysis. Among these clinical characteristics, only age at diagnosis, lymphovascular invasion, and clinical stage had a P value less than 0.05 by Log-rank test (Table 1). Multivariate Cox regression analysis of these 3 factors revealed that all of them were independent prognostic factors (Table 1). The results of this preliminary assessment demonstrated that the survival data in the TCGA UBC cohort, despite the censored data, were informative and applicable to identifying a prognostic signature.
WPI distributions and stratifications of UBC patients of clinical signature (A), RNA signature (B) and clinical-RNA signature (C), respectively.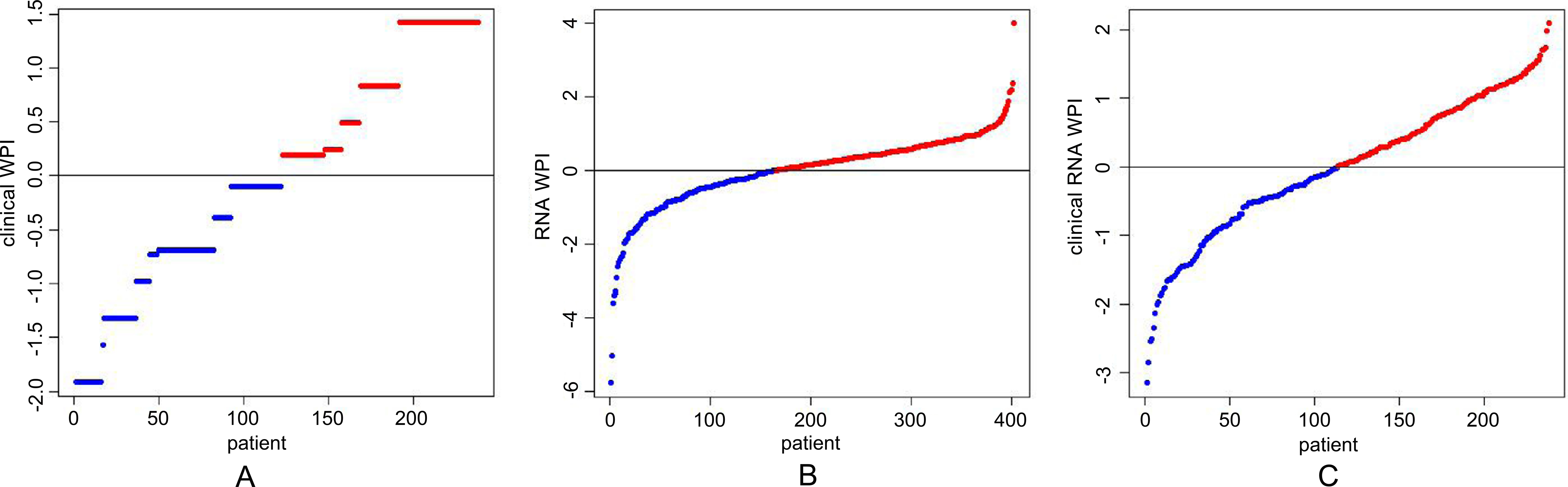
Kaplan-Meier plots for stratifications based on clinical signature (A), RNA signature (C) and clinical-RNA signature (E), respectively. ROC for WPIs based on clinical signature (B), RNA signature (D) and clinical-RNA signature (F), respectively.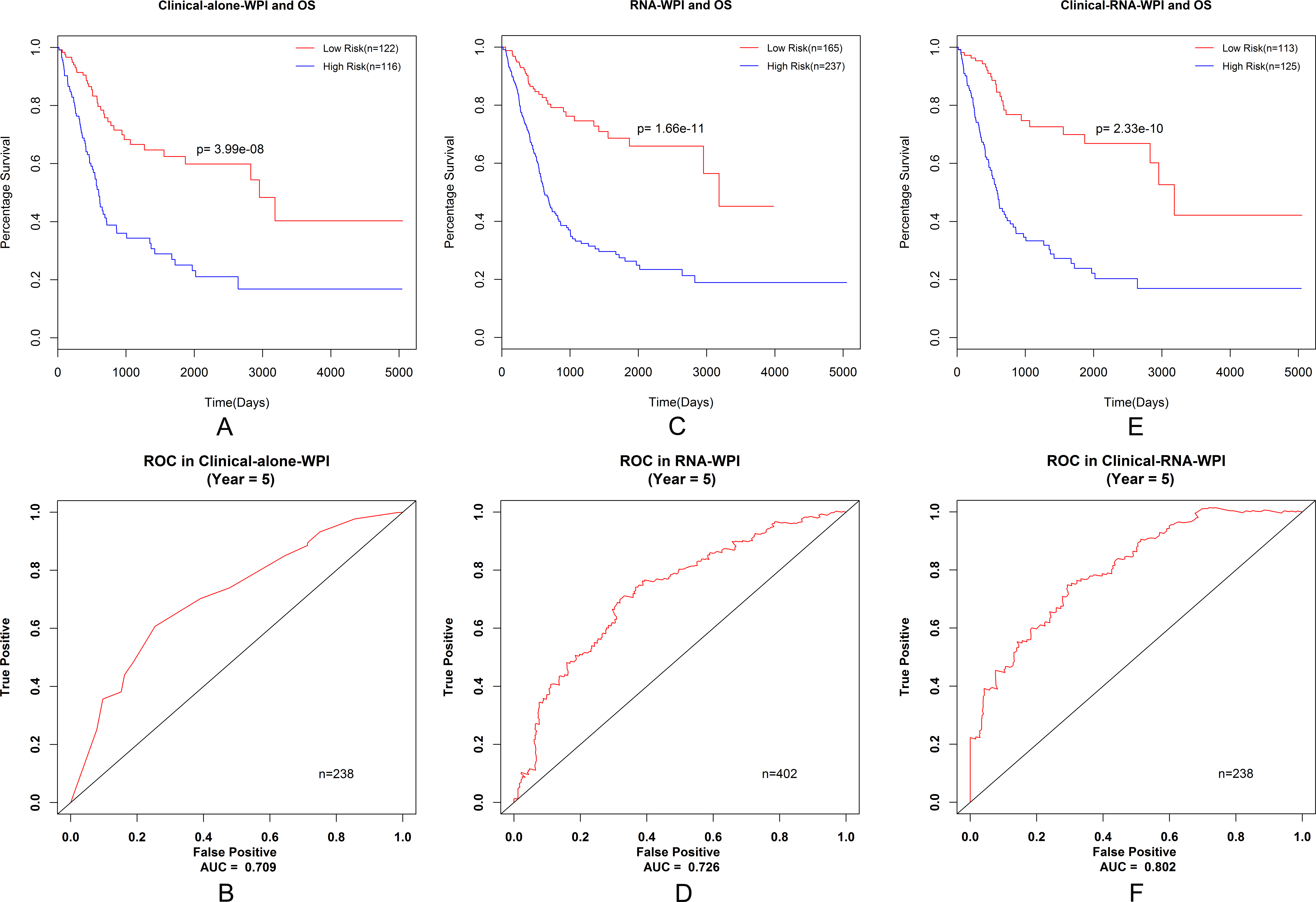
Slim gene ontology enrichment
Differential expression analysis revealed that 3043 RNAs were differentially expressed between UBCs and normal samples. Among these RNAs, 211 RNAs (adjusted P value
The WPIs ranged from
In all of the survival analyses, fewer events occurred after 5 years, we tested the ability of the integrated RNA signature to predict the survival outcome of UBC patients at, and around, this time point by time dependent ROC analysis. The AUCs were 0.709, 0.726 and 0.802 at 5 years of OS for clinical-signature (Fig. 3B), RNA-signature (Fig. 3D) and clinical-RNA signature (Fig. 3F), respectively. Thus, the integrated clinical-RNA signature had superior performance compared with clinical alone and RNA alone signatures whatever in terms to greater HR or greater AUC.
Network of proteins. A. The network predicted with methods of neighborhood, gene fusion, co-occurrence, co-expression, experiments, databases and text-mining. This network was enriched in interaction with a p-value of 4.22e-15. B. The network predicted only with experiments. Disconnected nodes were hidden. This network was enriched in interaction with a p-value of 0.0324.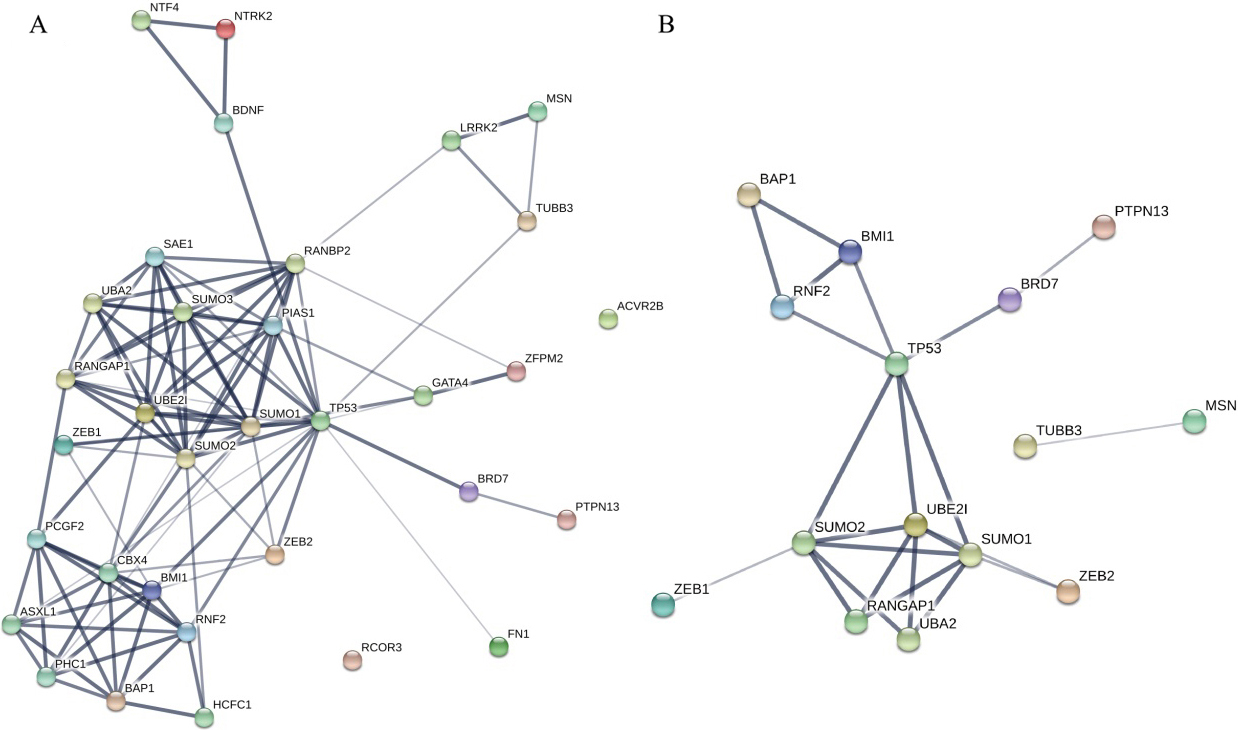
KEGG pathway enrichment
STRING database (
In cellular component analysis, we find that many gene products locate in chromosome and involve in regulating target genes by chromosome modification (Table 4). These gene products generally constitute some protein complex to bind chromatin.
In molecular function analysis (Table 4), molecular function of SUMO transferase activity is a major function in UBC patients. The enrichment of molecular function supports biological process. Besides, we can find that many genes are involved in protein binding, chromatin binding and domain binding. The main functions of these genes were binding and activity. The results indicated that these genes as direct regulators for regulation gene expression.
In KEGG pathway analysis, miRNA’s target genes and mRNA are mainly enriched in four KEGG pathways (FDR
Network analysis
STRING was used to visualize the protein-protein interaction network among miRNAs’ targets and mRNAs of the RNA signature. Figure 4A showed the network with methods of neighborhood, gene fusion, co-occurrence, co-expression, experiments, databases and text-mining. This network wags enriched in interaction, which assumed that these protein worked altogether and joined in the related pathways. After removing evidences with lower confidence and only using data experiments, a network with fewer genes was built (Fig. 4B). In both networks, TP53 was the center and had high degrees, indicating the important role of it in the prognosis of UBC.
Discussion
In this study, we proposed a novel integrated mRNA, miRNA and clinical signature that could predict UBC patient survival more accurate than clinical-only and RNAs-only signatures. Pathway analysis revealed that genes in our signature were involved in cell death and survival. All results suggested that the integrated signature was suitable and had superior prognostic value for UBC patient prognosis. Our study provides novel insights into the significance of bringing molecular and clinical information together for UBC prognosis.
Although markers for classifying UBC molecular subtypes have been identified, markers associated mainly with the pathogenesis of UBC may not predict the prognosis [32]. Furthermore, it is impertinent to use the clinical characteristics alone to distinguish the high-risk patients due to the underlying molecular heterogeneity within the clinically homogeneous tumor groupings. Thus we objected to identify common RNAs that consistently drive the outcome of UBC patients irrespective of the clinical or molecular subtypes. For UBC prognostic markers discovery, studies have been focused on either mRNA [9, 33] or miRNA profiles [7, 10, 11, 12, 13, 14] with limited sample size. To our knowledge, there is no study have been proposed a combination signature of mRNA expression, miRNA expression, and clinical characteristics to improve the UBC patient prognosis.
Of the prognostic RNAs, hsa-mir-200c was previously reported to be associated with UBC patient survival [34]. It has been reported that hsa-mir-200c regulates epithelial to mesenchymal transition (EMT) and restores expression of E-cadherin in breast and ovarian cancer cells [35, 36, 37]. Yu et al. reported that high levels of hsa-mir-200c expression inhibit cancer invasion and stimulate cancer cell proliferation, possibly via up-regulation of E-cadherin, and that high levels of hsa-mir-200c expression correlate with better survival of patients with curative resection of pancreatic cancer [38]. Furthermore, to the best of our knowledge, we are the first to report the hsa-mir-598, UBD, CATSPER2 and ZNF600 were also associated with UBC patient survival. Expression of miR-598-3p was down-regulated in the serum of breast cancer patients, using miR-598-3p as a biomarker, the sensitivity and specificity for the detection of breast cancer was 95.0 and 87.5% [39]. High expression of UBD correlates with epirubicin resistance and indicates poor prognosis in triple-negative breast cancer [40]. ZNF600 were down regulated in smokers compared to non-smokers [41], and cigarette smoking was an important risk factor for bladder cancer in both sexes [42]. Although the specific function of ZNF600 is unclear, ZNF600 expression is closely associated with smoking. Consistent with the GO result, UBD is not only an ubiquitin protein associated with ubiquitylation, which plays a vital role in survival [43]. CATSPER2 plays a central role in calcium-dependent physiological responses essential for successful fertilization, such as sperm hyperactivation, acrosome reaction and chemotaxis towards the oocyte. The relationship between prognosis of UBC and CATSPER2 expression has not been reported yet. ZNF600 belongs to the krueppel C2H2-type zinc-finger protein family which involved in transcriptional regulation. Transcriptional regulation factor such as ZNF600 generally affects cancer by promoting cell proliferation.
Among the risky genes, hsa-mir-143, RUNX2,LAMA2, INHBB, KCNK6, PLSCR4, PAM andPDGFD are associated with tumor development. In previous report of study, microRNA-143 as a tumor suppressor for bladder cancer and microRNA-143 inhibits cell migration and invasion by targeting matrix metalloproteinase 13 in prostate cancer. Thus, the function of microRNA-143 is heterogeneous in different study. And this condition needs to be further study. RUNX2 and TP53 might be functionally related and are likely involved in bladder tumor carcinogenesis and aggressiveness, and RUNX2 and p53 independently predict early tumor recurrence in bladder carcinoma patients, with the highest prediction accuracy being achieved on their combined high expression [44]. TP53-induced miR-34a contributes liver regeneration termination via regulation of INHBB and MET. Other risky genes have not been reported in bladder cancer yet. However, LAMA2, INHBB and PDGFD all involved in cell migration and embryonic development [45, 46]. In fact, cell migration is closely associated with cancer. On the other hand, embryonic development and cancer development have some similar biological processing such as rapid cell proliferation and EMT pathway. KCNK6, PLSCR4 and PAM performed their special role in bladder cancer. Although these three genes are high-risk genes in bladder cancer, it is difficult to elaborate their functions. According to main functions of PLSCR4 and PAM, they are ion binding proteins. And KCNK6 is a transport for K
In conclusion, we have identified a 3-clinical and 13-RNA integrated signature that can predict the survival outcome of UBC patients more accurate than RNA-alone or clinical-alone signatures. Our findings may help researchers understanding of UBC cell death and survival, develop targeted therapy, and identify high-risk UBC patients for better disease management.
Footnotes
Acknowledgments
which made the genomic data of UBC available. We also thank the anonymous reviewers for helpful comments. This work was supported by The Postdoctoral Science Foundation of Central South University.
Conflict of interest
The authors have declared that no competing interests exist.
Supplement material
Mature sequences of miRNA signature and experimentally validated mRNA targets
Mature miRNA
Targets
Mature miRNA
Targets
hsa-mir-143-3p
KRAS
hsa-miR-200c-3p
TUBB3
MYO6
BMI1
DNMT3A
SIP1
FNDC3B
BAP1
MAPK7
ZEB2
FSCN1
ZEB1
HK2
Zeb2
SERPINE1
Zeb1
MACC1
FN1
PTGS2
ZFPM2
JAG1
UBE2I
AKT1
PTPN13
COL1A1
RNF2
HRAS
RCOR3
MDM2
BRD7
BCL2
ACVR2B
MMP13
MSN
SDC1
NTRK2
RREB1
ERRFI1
CD44
CCNE2
KLF5
XIAP
BRAF
BCL2
TNF
TIMP2
NR2C2
FBLN5
Supplementary Table 1, continued
Mature miRNA
Targets
Mature miRNA
Targets
IL13RA1
VEGFA
DYT10
NCAM1
COL3A1
Vldlr
DDX6
Reln
LIMK1
IKBKB
HNF4A
FLT1
KLF9
hsa-miR-143-5p
COX2
hsa-miR-200c-3p
TBK1
IGF1R
PMAIP1
NTF3
LPAR1
EDNRA
RHOA
KLHL20
PTPRD
ELMO2
ERBB2IP
WDR37
VAC14
TCF7L1
RASSF2
HOXB5
RIN2
KLF11
SEPT7
SHC1
MYB
ETS1
USP25
EFNA1
RND3
DNMT3A
DNMT3B
SP1
CFL2
CDH11
SEC23A
KDR
HFE
DLC1
ATRX
ZNF217
BTC
ZFPM1
PIN1
KRAS
NOTCH1
GATA4
SUZ12
ROCK2
UBQLN1
E2F3