
Abstract
Background:
Microbiome dysbiosis plays a role in the pathogenesis of many urological diseases, including bladder cancer (BC). The aim of the study was to compare the urinary and gut microbiota of patients with BC with a healthy control (HC) group.
Methods:
The study group included patients hospitalized in 2020 to 2021 with diagnosed BC and HC. Prior to the transurethral resection of bladder tumor, patients collected their urine and stool which was then subjected to 16S rRNA gene sequencing.
Results:
Overall, 25 patients were enrolled in the study: 18 in the BC group and 7 in the HC group. Analysis of the urine and stool microbiome showed no statistically significant differences between patients with BC and HC in alpha diversity, beta diversity, and difference in taxa relative abundance. Detailed analysis of urine and stool microbiome depending on patient- and tumor-related characteristics also showed no statistically significant differences in alpha diversity and beta diversity. Differences in abundance (ANCOM) were noted in both types of samples in patients with BC. In the urine test, genus Lactobacillus was more common in patients with a positive history of Bacillus Calmette–Guérin (BCG) therapy, while genus Howardella and the strain Streptococcus anginosus were more common in women. In stool samples, abundance of phylum Desulfobacterota was most abundant in Grade G1 and least in G2. Class Alphaproteobacteria, order Rhodospirillales, order Flavobacteriales, and family Flavobacteriaceae were more common in women.
Conclusions:
The microbiome of urine and stool of patients with BC does not differ significantly from that of HC; however, its composition in patients with BC varies according to the patient’s sex.
Introduction
Bladder cancer is one of the most frequently diagnosed cancers in the world population. It affects nearly 500 000 people each year, and more than 1/3 of them die from the disease. 1
The cause of bladder cancer is multifactorial but not fully understood. The best documented risk factors include smoking, occupational exposure to amines and aromatic hydrocarbons, administration of cyclophosphamide or pioglitazone, and history of radiotherapy. 2 Researchers are becoming more and more aware that in addition to environmental and genetic factors, the human microbiome can also contribute to the pathogenesis of many chronic diseases, including cancer.3-5
Advances in DNA sequencing, among others, allowing for high-throughput microbiota testing, have shown that the bladder has its own endogenous microbiome, and the dogma that urine must be sterile in healthy individuals has been overthrown.3,4,6-9 Further studies suggest that microbiota dysbiosis plays a role in the pathogenesis of many urological diseases, such as interstitial cystitis, neurogenic bladder, overactive bladder, urge incontinence, benign prostatic hyperplasia, and chronic prostatitis.3,6,8-10
The relationship between chronic inflammation, microbiome, and solid tumors has been confirmed in many cancers, including prostate cancer.5,6 The participation of the microbiome in the pathogenesis of bladder cancer is also suspected.3-9 However, the available data are insufficient and further research is needed.
The aim of this study was to compare the urinary and fecal gut microbiota of patients with bladder cancer with a healthy control group. In addition, we assessed whether there is an association between the composition of the urinary and gut microbiome of patients with bladder cancer and patient- and tumor-related characteristics.
To our knowledge, this is the first study to analyze both the urine and gut microbiome in the same group of patients, and the first to analyze the fecal microbiome in bladder cancer patients according to patient- and tumor-related characteristics.
Materials and Methods
The study was carried out as part of the grant “Młodzi Naukowcy” (STM.C090.20.077) and was approved by the Bioethics Committee of the Wrocław Medical University (197/2020).
Study design and specimen collection
The study group included patients hospitalized at the University Center of Excellence in Urology in 2020 to 2021 with diagnosed primary or recurrent bladder cancer. The cancer was suspected on the basis of an interview, physical examination, and ultrasound (USG) examination, and the diagnosis was confirmed by histopathologic examination after transurethral resection of the bladder tumor (TURB). The control group included patients hospitalized in the same period of time for other indications, in whom the presence of bladder cancer was excluded by means of USG examination. After giving informed written consent to participate in the study, patients completed a questionnaire collecting information on sociodemographic characteristics.
For microbiome analysis, patients were asked to collect urine and stool. Urine was collected from the midstream in accordance with generally accepted urological standards under the supervision of a urological nurse. Stool was collected using a Kałszyk stool sample collection kit (Konrad Kosowski, Wąchock, Poland), which consists of biodegradable paper applied to the toilet to collect stool and prevent contamination and a sterile container with a spatula. The bowl-shaped paper stool collector is placed on the toilet and the patient defecates into it. Then, using a spatula, the patient collects the stool into the plastic container provided in the kit, so that it is half-filled. In both groups, urine and stool were collected after the USG examination, and in the case of the research group, before the TURB. After collection, the biological material was stored at −80°C until DNA isolation.
DNA isolation
DNA from urine was isolated with the use of the QIAamp DNA Micro Kit (Cat. no. 56304, QIAGEN) according to the manufacturer’s instructions. Briefly, 10 mL of urine was centrifuged (6000g, 2 min, RT), and the obtained pellet was washed once in AE buffer and dissolved in ATL lysis buffer with the addition of proteinase K and β-mercaptoethanol. After 1-hour incubation (56°C with shaking), buffer AL and 96% ethanol were added, and the obtained suspension was vortexed and transferred to the QIAamp MinElute column. Next, a series of washing steps were performed with AW1 and AW2 buffers. Finally, 25 μL of AE buffer was added to elute the DNA from the column. For better purification, buffer AL was supplemented with carrier RNA (included in the kit).
For DNA isolation from feces, QIAamp® PowerFecal® Pro-DNA Kit (Cat. no. 51804, QIAGEN) was used. In short, approximately 250 mg of stool was added to the PowerBead Pro Tube together with Solution CD1 and the sample was homogenized horizontally at 1400 r/min (Thermomixer; Eppendorf). The homogenate was later centrifuged (15 000g, 1 min) and the supernatant was moved to a clean tube first with Solution CD2 and next with Solution CD3. The obtained lysate was moved to the MB Spin Column, and a series of centrifugation and washing steps were performed with Solution EA and Solution C5. Finally, 50 μL of Solution C6 was added on top of the column and isolated DNA was centrifuged (15 000g, 1 min) to a new 1.5 mL tube.
DNA library preparation and sequencing
Total DNA extracts were subjected to the generation of 16S rRNA gene amplicon libraries, which were prepared using the QIAseq 16S/ITS Region Panels (QIAGEN) for the V3 to V4 region, according to a standard protocol.
Paired-end sequencing (2 × 276) was conducted using MiSeq Reagent Kits v3 (600 cycles) on MiSeq (Illumina).
The DNA libraries for next-generation sequencing (NGS) were prepared using the QIAseq 16S/ITS Panel kit, for the V3 to V4 variable region, according to a standard protocol. This kit employs a 2-stage process using polymerase chain reaction (PCR) reactions. The variable regions are amplified in the first PCR reaction, where phased primers (that add 0 to 11 additional bases before the target-specific primer) are used to solve the problem of low library diversity. The next step is the purification of the amplified products on magnetic beads, which allows for the separation of PCR products from the remaining components of the reaction mixture. Then, in the second PCR reaction, adapters are added to the amplicons, which add a 2 × 8-nucleotide index sequence (indexes at the 3’ and 5’ ends) that allow to identify to which sample a given read belongs, and allow to bind to the flow cell, on which the amplification reaction occurs during sequencing. The obtained product is again subjected to purification using magnetic beads, resulting in the acquisition of DNA libraries, which are then subjected to qualitative and quantitative analysis.
Quantitative analysis was conducted by measuring the concentration of the obtained libraries. Libraries were measured using the QuantiFluor® dsDNA System kit (Promega) on a Quantus™ Fluorometer. For the qualitative analysis, we used electrophoresis to verify the presence of DNA fragments of the intended size. This process was conducted using a TapeStation device (Agilent), with High Sensitivity D1000 ScreenTape. This automatic system is intended for the analysis of DNA particles ranging in size from 35 to 1000 bp. Samples that reached a minimum input concentration of 2 nM were sequenced.
Bioinformatic analysis
The QIIME2 2021.8 and accompanying plugins were employed for bioinformatic analysis of microbiome data. 11
Custom script incorporating Cutadapt was used for phased primer removal and demultiplexing. 12 The demux plugin’s summary method assessed read quality, while the dada2 plugin processed paired-end reads through trimming, denoising, dereplication, and chimera filtering. 13
These operations allowed for the identification of every observed amplicon sequence variant (ASV), also referred to as Operational Taxonomic Units (OTUs), with a 100% identification rate.
The q2-phylogeny plugin’s align-to-tree-mafft-fasttree routine performed multiple sequence alignment using the mafft method, masked highly variable positions, and constructed phylogenetic trees with fasttree.14,15
Samples were rarefied to 12 625 per sample (subsampled without replacement, to minimal number of sequences in a batch).
The process of the rarefaction was used to standardize the number of sequences across different samples to enable unbiased comparison. This process is important because different samples can yield different numbers of sequences, which can introduce bias when comparing the richness or diversity of the microbiomes.
The q2-diversity plugin estimated alpha (diversity within a particular sample) and beta diversities (the diversity of species between different samples) and conducted principal coordinate analysis (PCoA).
Principal coordinate analysis is a method used to visualize and explore similarities or dissimilarities in multi-dimensional data, such as microbiomes, by plotting objects in a low-dimensional space, here 2-dimensional. The distances between points represent their dissimilarities. This enables the identification of clusters, trends, and relationships.
The naïve Bayesian classifier, trained on 16S rRNA gene sequence fragments from the SILVA 138 SSURef NR99 database, was employed using the fit-classifier-naive-Bayes method from the feature-classifier plugin to assign taxonomy to ASVs.16-18
To investigate whether the microbiomes across groups differ in the abundance of specific bacterial taxa, the analysis of the composition of microbiomes (ANCOM) method was applied, as implemented in the q2-composition plugin with default parameters. 19
Descriptive statistics were presented as means and standard deviations. Parameter differences between groups were analyzed using the Kruskal–Wallis test. Beta-diversity group significance testing involved permutational multivariate ANOVA (PERMANOVA) with 999 permutations. Statistical significance was determined by P-values less than .05.
Results
Sequencing data, after initial processing, ie, extraction of reads for the V3 to V4 variable region, are available in the NCBI BioProject database (PRJNA1017542).
Patient characteristics
Biological material was collected from 30 patients. Of these, 25 patients were included in the analysis of the urinary microbiome, including 18 patients with bladder cancer and 7 healthy patients. However, 5 patients were excluded from the urinary microbiome analysis due to insufficient genetic material for sequencing. In total, 30 patients were included in the gut microbiome analysis, including 23 with bladder cancer and 7 healthy patients.
The majority of patients (>73%) were men. The average age was 73 years. In addition, a significant proportion (>60%) of patients was active or former smokers. The most common stage of advancement was Ta low-grade tumors and more often they were recurrent tumors. Most patients did not receive Bacillus Calmette–Guérin (BCG) instillations.
Detailed characteristics of patients with bladder cancer are presented in Table 1.
Bladder cancer patient characteristics.

Abbreviations: BCG, Bacillus Calmette–Guérin; BMI, body mass index; TURB, transurethral resection of the bladder tumor.
Relative frequency
Figures 1 and 2 show the relative abundance of the microorganisms at the phylum level in the individual urine and stool samples.

Relative abundance at phylum level—urine samples. BC indicates bladder cancer patients; HC, healthy controls.

Relative abundance at phylum level—stool samples. BC indicates bladder cancer patients; HC, healthy controls.
Urinary microbiome
Comparison of patients with bladder cancer with a control group
No statistically significant differences in the urinary microbiota were observed between the bladder cancer group and the healthy controls at the alpha-diversity level (Table 2, Figure 3).
Urine and stool microbiome alpha-diversity comparison.

Abbreviations: BC, bladder cancer patients; HC, healthy controls.
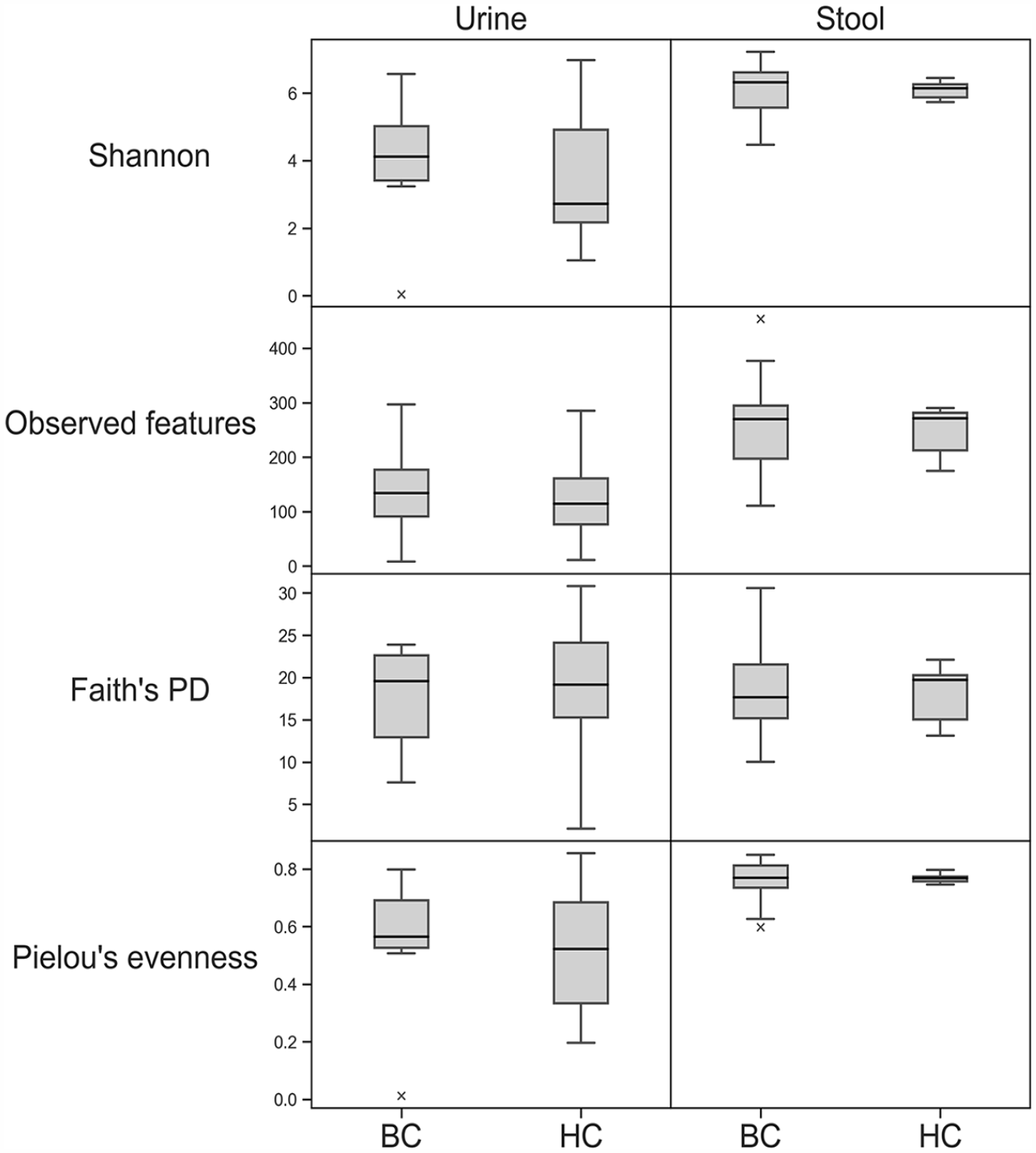
Urine and gut microbiome alpha-diversity comparison. BC indicates bladder cancer patients; HC, healthy controls; PD, phylogenetic diversity.
Also, no statistically significant differences between the groups were found in beta diversity, in all 4 metrics checked (Jaccard, Bray–Curtis, Unweighted UniFrac, and Weighted UniFrac) (Figure 4).

Urine and gut microbiome beta-diversity comparison. (A) Jaccard. (B) Bray–Curtis. (C) Unweighted UniFrac. (D) Weighted UniFrac. BC indicates bladder cancer patients; HC, healthy controls.
The analysis of composition of microbiomes, the method ANCOM revealed, that also in the terms of abundance at any of the 6 taxonomic levels (from phylum to genus) of urine samples from patients with bladder cancer does not differ from control. 19
Detailed analysis of the microbiome of patients with bladder cancer
Detailed analysis of the urinary microbiome according to patient- and tumor-related factors, such as sex, BMI, age, nicotinism history, T stage, grade, receiving BCG therapy, and primary/recurrent tumor, showed no statistically significant differences in either both alpha and beta diversity (Tables 3 and 4).
Alpha-diversity significance among stool samples of bladder cancer patients.

Abbreviation: BCG, Bacillus Calmette–Guérin; HG, high grade; LG, low grade.
Beta-diversity significance among urine and stool samples of bladder cancer patients.
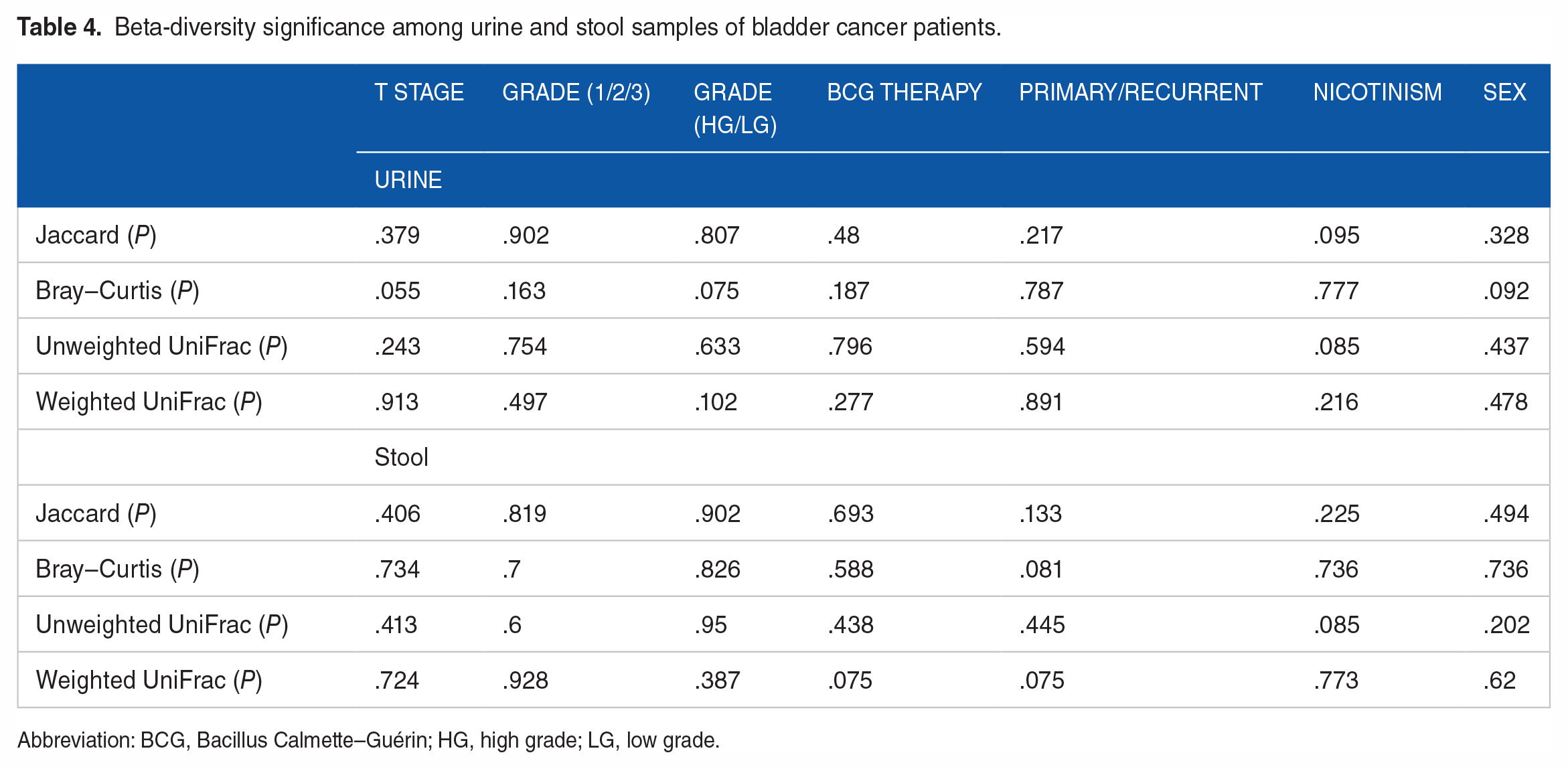
Abbreviation: BCG, Bacillus Calmette–Guérin; HG, high grade; LG, low grade.
The analysis of the composition of microbiomes showed differences between urine samples among patients with bladder cancer. Genus Lactobacillus was more common in patients with a positive history of BCG therapy, while genus Howardella and the strain Streptococcus anginosus were more common in female patients (Figure 5). This result should be interpreted with caution, as the resolution of the method does not allow for certainty down to the strain level.

Detailed ANCOM analysis. ANCOM indicates analysis of the composition of microbiomes; F, female; M, male, BCG, Bacillus Calmette–Guérin.
Gut microbiome
Comparison of patients with bladder cancer with a control group
Comparison the fecal microbiota of bladder cancer patients to controls showed that there were no differences in either the alpha- (Table 2) or beta-diversity levels (Figure 4). The analysis of the composition of microbiomes method also did not show bacterial taxa that could be differentially abundant across the 2 groups.
Detailed analysis of the microbiome of patients with bladder cancer
Detailed analysis of the gut microbiome according to patient- and tumor-related factors, such as sex, BMI, age, nicotinism history, T stage, grade, receiving BCG therapy, and primary/recurrent tumor, showed no statistically significant differences in either both alpha- and beta-diversity levels (Tables 3 and 4).
The analysis of the composition of microbiomes analysis showed differences between stool samples among patients with bladder cancer. Abundance of phylum Desulfobacterota differed across tumor grade, it was most abundant in Grade G1 and least in G2. Class Alphaproteobacteria, order Rhodospirillales, order Flavobacteriales, and family Flavobacteriaceae were more common in female patients (Figure 5).
Discussion
Until recently, it was believed that the urine of healthy people should be sterile. Advances in microbiological diagnostic techniques and the use of 16S rRNA amplicon sequencing have allowed this dogma to be overthrown. Further research allowed to characterize the urine microbiome of healthy people. 20 Based on the evidence indicating the role of the gut microbiome in the pathogenesis of various diseases, research has been initiated to determine whether such a relationship exists between the urobiome and urological diseases, including cancer. One of the urological malignancies in which the role of the urinary microbiome is postulated is bladder cancer. Owing to the limited amount of research on this topic, we focused on this cancer in our study.
There are 2 hypotheses to explain dysbiosis in bladder cancer. According to the first, the non-physiological urinary microbiome is a risk factor and may promote and participate in the development of bladder cancer. The second suggests that the environment of the developing tumor leads to a change in the composition of urobiome. 21
However, in our study, we found no differences between the urinary microbiome of healthy individuals and patients with bladder cancer, both in alpha and beta diversity. Similar results were obtained in the studies by Bučević Popović et al, 6 Pederzoli et al, 22 and Moynihan et al. 23 Yet, the findings of other researchers do not support this evidence. Hussein et al, Bi et al, Qiu et al, and Ma et al showed differences in diversity between the groups.9,24-26 Wu et al and Zeng et al noticed a greater richness of the urinary microbiome in patients with bladder cancer.7,27 Opposite results were obtained by Chipollini et al, who observed a lower richness and evenness of the urobiome of patients with bladder cancer. They postulate a hypothesis in which the urinary microbiome is rich in healthy people, while in the neoplastic process, 1 microbial community dominates. 28 Researchers have tried to determine the predominant microbes found in the urine of patients with bladder cancer, but the results are inconsistent. Greater abundance of the genus Fusobacterium, 6 Streptococcus 4 , or Actinomyces,9,24 especially Actinomyces europaeus 9 has been described. Our ANCOM analysis, however, showed no statistically significant differences in the prevalence of microorganisms at any of the 6 taxonomic levels (from phylum to genus).
It is suggested that differences in the characteristics of the urinary microbiome between different stages of bladder cancer could be used as a biomarker to stratify the risk of progression and recurrence. In muscle-invasive bladder cancer, Chipollini et al described higher abundances of the genera Bacteroides and Faecalibacterium, and Hussein et al described the genera Haemophilus and Veillonella.24,28 In addition, patients with high-risk non–muscle invasive bladder cancer showed increased richness in the urine microbiome, 7 and patients with recurrent bladder cancer had greater urobiome alpha diversity.26,27 In our studies, however, we did not show any differences in alpha and beta diversity depending on the stage of the cancer or its nature (primary/recurrent).
The incidence of bladder cancer varies by sex, with a higher incidence in men. This was thought to be mainly due to the higher prevalence of smoking in the male population. Over the years, the number of smoking men and women has changed, without a significant impact on the incidence of bladder cancer. 29 For this reason, the search for other factors influencing the difference in the incidence of bladder cancer between women and men began. The difference in the urine microbiome of men and women is believed to be the likely responsible factor. 24 In our study, we found no differences in the alpha and beta diversity of the urine microbiome of patients with bladder cancer depending on sex; however, the Howardella genus and the Streptococcus anginosus strain were more common in women. Similarly, Hussein et al showed no differences in the diversity of urine between sexes; however, in the urine of women, there was a higher abundance of the genera Lactobacillus, Actinotignum, Prevotella, Veillonella, and Campylobacter. 24 Pederzoli et al described a higher incidence of the genus Klebsiella in the urine of female patients with bladder cancer. 22 Owing to the small study groups and the prevalence of studies involving only male patients, one should be careful in drawing conclusions from the results obtained so far. More studies with more patients of both sexes are needed.
Bacillus Calmette–Guérin instillations are one of the treatments for patients with high-risk bladder cancer. 2 It is suspected that BCG instillations may interact with the urinary microbiome, influencing its composition, or the urinary microbiome may affect the effectiveness of response to BCG therapy. 30 In our study, the genus Lactobacillus was more frequently found in the urine of patients with a history of BCG therapy. Owing to the small group of patients, we did not analyze the response to treatment. It is believed that Lactobacillus may compete with BCG in binding to fibronectin, reducing its effectiveness. 31 However, Sweis et al showed that Lactobacillales were more abundant in the urine of patients undergoing BCG therapy without recurrence, and the order Enterobacterales was more abundant in the urine of patients with recurrence.32,33 In another study, Hussein et al showed a high incidence of the genera Serratia, Brochothrix, Negativicoccus, Escherichia-Shigella, and Pseudomonas in the urine of BCG-responsive patients. 24
About 99% of the human microbiome is found in the intestinal tract. Dysbiosis of the gut microbiome leads to the development of numerous diseases, not only locally but also outside the digestive tract. 34 Its role in the pathogenesis of cancers, including urological ones, is being studied more and more often. Recent studies have shown a connection between the gut microbiome and the development of prostate cancer. 35 It is believed that such a relationship may also occur in the case of bladder cancer. So far, only a few studies have been conducted on this topic. He et al showed a reduced abundance of Clostridium cluster XI and Prevotella in the gut microbiome of patients with bladder cancer. 34 Different results were obtained by Bukavina et al, who revealed an increased abundance of Prevotella, along with Fusobacteria and Bacteroides. 36 Qin et al and He et al found decreased abundance of Clostridiales bacterium in the stools of bladder cancer patients, but increased Streptococcus and Escherichia. In addition, they found a reduction in probiotics in bladder cancer patients, such as Lactobacillus, Bifidobacterium, and Butyrate-producing bacterium, which are believed to be crucial in protecting the body and inhibiting tumor growth. 37 In our study, we found no differences in the gut microbiome between bladder cancer patients and healthy controls, but the ANCOM analysis showed a difference between bladder cancer patients by sex and tumor grade. However, due to the small number of groups, further research is needed.
It is believed that the gut microbiome plays a role not only in the pathogenesis of bladder cancer but also in its response to systemic treatment. Bukavina et al assessed the role of the gut microbiome in the chemoresponse to neoadjuvant chemotherapy in patients with bladder cancer before radical cystectomy. Overall, there were no significant differences in alpha and beta diversity between chemotherapy responders and non-responders. However, an association was found between the higher frequency of Bacteroides during chemotherapy and the presence of residual disease at the time of radical cystectomy, regardless of the chemotherapy regimen. 36
Pederzoli et al instead, assessed the role of the gut microbiome on the response to neoadjuvant immunotherapy before radical cystectomy. Alpha diversity was higher in immunoresponders than in non-responders. There was no difference in beta diversity, but the genus Sutterella was enriched in responders and the species Ruminococcus bromii was enriched in non-responders. 38
In our work, we showed differences in the gut microbiome of patients with bladder cancer depending on the tumor grade and the patient’s sex. To our knowledge, this is the first study to compare the gut microbiome of patients with bladder cancer according to patient- and tumor-related characteristics. We showed a higher incidence of class Alphaproteobacteria, order Rhodospirillales, order Flavobacteriales, and family Flavobacteriaceae in female with bladder cancer. In addition, we detected differences in the abundance of phylum Desulfobacterota across tumor grade, which was most abundant in Grade G1 and least in G2. Owing to the size of the study group and the lack of other literature, the results should be treated as preliminary and further research is necessary.
Our work has several limitations. First, the study groups were small and single-centered due to the cost of sequencing. Second, these groups were very diverse in sociodemographic and tumor-related factors, such as history of BCG therapy or nicotinism. For both of these reasons, the results should be interpreted with caution and treated as preliminary. We hope for a multicenter study in the future. Third, urine in both groups was collected from the midstream, which could be contaminated with microbiome in the area of the urethral opening. Hourigan et al showed no differences in the urine microbiome collected from midstream and cystoscopy, only in the sex-specific analysis, differences were found in men. 10 In addition, catheterization or cystoscopy carry the risk of urethral trauma or infection, therefore performing them in healthy people raises some ethical concerns. What is more, the study evaluated the biological material collected on the day before resection, without re-evaluation at a later time and assessment of response to treatment and the presence of recurrence. A further prospective study is planned that will take these aspects into account. Another limitation results from the technique of 16S rRNA sequencing, which does not allow the detection of microorganisms other than bacteria and archea, such as fungi or viruses, which may play a role in the occurrence of the disease.
Conclusions
In conclusion, we compared the urine and feces microbiota of patients with bladder cancer and healthy patients and did not show significant differences, which does not contradict the hypothesis about the role of the microbiome in the pathogenesis of bladder cancer. However, detailed analysis showed differences in the microbiome between sexes and depending on tumor-related characteristics, such as stage and history of BCG therapy. These preliminary results in a small number of patients open the door to further studies that may confirm these relationships. Owing to the large discrepancies between the studies, it seems necessary to develop a consensus on the technique of collecting urine, the schedule of collections, and the period of observation. The results obtained from the multicenter prospective study will allow for a better understanding of the pathogenesis of bladder cancer and the use of the microbiome as a diagnostic or risk stratification tool. Therapies modifying the microbiome may in the future allow for a better response to current treatment or reduce the risk of progression and recurrence of cancer. Accurate research of the microbiome, both gut and urinary, will allow for a more personalized approach to the patient and this disease.
Footnotes
Author Contributions
WK and JC contributed to the conception and design. BM and TS involved in administrative support and provision of study materials or patients. LN and KB contributed in collection and assembly of data. JC, PZ-R, LL, KP-P, and SG participated in data analysis and interpretation. All authors participated in article writing and approved the final article.
Ethics Approval and Consent to Participate
The study was approved by the Bioethics Committee of the Wrocław Medical University (197/2020). All patients gave written informed consent to participate in the study.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was financed by the grant “Młodzi Naukowcy” (STM.C090.20.077).
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.