
Abstract
Metabolism of neoplastic cells is shifted toward high glucose uptake and enhanced lactate production. Lactate dehydrogenase (LDH), which is comprised of two major subunits, LDH-A and LDH-B, reversibly catalyzes the conversion of pyruvate to lactate or lactate to pyruvate. LDH-A has a higher affinity for pyruvate and is a key enzyme in the glycolytic pathway. Elevated LDH is a negative prognostic biomarker not only because it is a key enzyme involved in cancer metabolism, but also because it allows neoplastic cells to suppress and evade the immune system by altering the tumor microenvironment. LDH-A alters the tumor microenvironment via increased production of lactate. This leads to enhancement of immune-suppressive cells, such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and dendritic cells (DCs); and inhibition of cytolytic cells, such as natural killer (NK) cells and cytotoxic T-lymphocytes (CTLs). By promoting immune-suppression in the tumor microenvironment, LDH-A is able to promote resistance to chemo/radio/targeted therapy. Here we discuss the evidence that LDH is both a metabolic and an immune surveillance prognostic biomarker and its elevation is harbinger of negative outcome in both solid and hematologic neoplasms.
Introduction
In many neoplastic cells, metabolism is shifted toward high glucose uptake and enhanced lactate production, regardless of oxygen availability. This phenomenon is known as the Warburg effect, and it is one of the fundamental metabolic rewiring processes that ensues during malignant transformation [1]. This occurs not because of the mitochondrial dysfunction, as was thought originally, but because of the dependency of cancer cells on the multiple metabolites of glucose involved in the synthesis of nucleic acids, fatty acids, and lactate to promote intracellular signaling, microenvironmental angiogenesis, and net tumor growth [2]. It is estimated that tumor cells utilize up to 30 times more glucose than normal cells and produce up to 40 times more lactic acid, irrespective of oxygen supply [3].
One of the key enzymes in the glycolytic pathway is lactate dehydrogenase (LDH), a tetrameric enzyme that catalyzes the reversible conversion of pyruvate to lactate, coupled with the oxidation of nicotinamide adenine dinucleotide dehydrogenase (NADH) to NAD
Elevated LDH is well established as a negative prognostic biomarker in many cancers and is directly correlated with increased tumor growth and proliferative index, maintenance, metastasis, and tumor survival. Here, we propose that elevated LDH conveys a poor prognosis not only because it is a key enzyme involved in cancer metabolism, but also because it alters the tumor microenvironment in hematologic and solid neoplasms, allowing tumor cells to suppress and evade the immune system.
Elevated serum LDH in immunosuppressed conditions
The link between elevated serum LDH and immunosuppression, independent of malignancy, has been suggested before. Studies have shown that there is a correlation between the incidence of opportunistic infections in patients infected with human immunodeficiency virus (HIV), CD4
The link between immunosuppression and malignancy has been studied extensively in patients who receive pharmacological immunosuppression for a variety of reasons such as solid organ transplantation and autoimmune diseases [7, 8]. Elevated LDH is a common laboratory finding in these patient populations and often is considered a negative risk factor for clinical outcomes [9, 10].
LDH is normally cleared by receptor-mediated endocytosis by macrophages in the liver, spleen and bone marrow [11]; however, when the immune system within the tumor microenvironment is altered during tumor growth, this may lead to insufficient elimination of LDH from tumor. These data suggest that in addition to increased production of LDH by neoplastic cells mainly due to Warburg effect, decreased destruction or clearance of LDH in immunocompromised environment may contribute to the high serum LDH level in these patients. Here, we propose that a serum LDH level above the upper limits of normal is not only a negative prognostic biomarker for malignant cells, but can also be a marker of immune suppression in host. The mechanisms involved in tumor-induced immunosuppression that are associated with both plasma and tumor high LDH levels are described below.
Elevated serum LDH is a negative prognostic biomarker for multiple hematologic and solid neoplasms
Total LDH has become a valuable prognostic bio- marker. Elevated serum LDH is a well-established strong negative predictor of survival in patients with Hodgkin’s and non-Hodgkin’s lymphomas. In one of the first studies investigating the prognostic effects of LDH, Garcia et al. [12] found that serum LDH levels
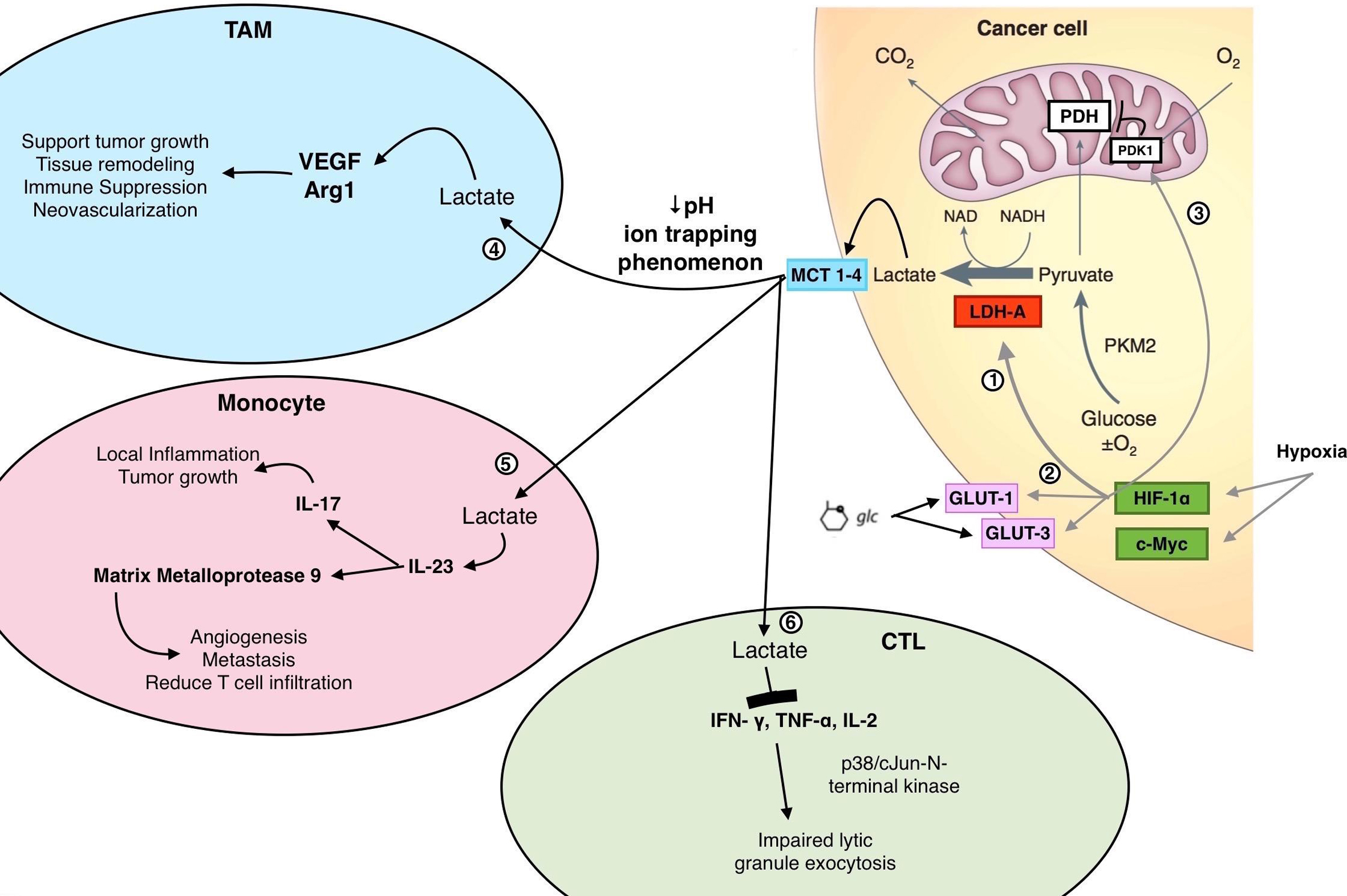
Neoplastic cell metabolism and resultant effects of LDH-A on immune cells within the tumor microenvironment. Within cancer cells, metabolism is shifted toward enhanced lactate production, regardless of oxygen availability (Warburg effect). Within a hypoxic environment, 1) the oncoproteins HIF-1
In a meta-analysis including 73 clinical studies, Petrelli et al. [16] showed that a high LDH is associated with poor survival in several solid tumors; this prognostic effect was most pronounced in renal cell carcinoma, as well as gastric, prostate, nasopharyngeal and lung cancers. Determination of elevated serum LDH was based on a predefined cut-off for serum LDH (median 254 U/L) in 34 out of these 73 clinical studies.
Similarly, in the prognostic staging system for cutaneous melanoma, elevated serum LDH above the upper limits of normal (ULN) at the time of staging was the most predictive independent factor of diminished survival [17], even after accounting for site and number of metastases. The degree of elevation of LDH above ULN also correlated with worse overall survival in patients with melanoma.
In a retrospective analysis of prognostic factors in patients with bone metastases from breast cancer, serum LDH above the ULN but less than two-times the ULN correlated with a two-fold increased risk of death; while serum LDH levels above two-times the ULN correlated with a six-fold increased risk of death [18].
Importantly, the degree of elevation of serum LDH has prognostic significance in men with germ cell tumors. The International Germ Cell Cancer Collaborative Group risk stratification system includes ß human chorionic gonadotropin (ß-hCG),
Overall, in clinical studies assay conditions in relation to serum LDH were not standardized, so there were no cut-off levels when relating LDH levels to prognosis of the many malignancies described above. Instead, the degree of elevation of LDH mentioned was in relationship to the upper limit of normal provided by each laboratory. There is variation in absolute reference ranges for the upper limit of normal across centers.
Role of LDH-A in tumor growth, maintenance and metastasis
Neoplastic cells have fundamentally enhanced production of lactic acid with preferentially high production of LDH isoenzymes rich in LDH-A. The expression of LDH-A is upregulated by the oncoproteins hypoxia-induced factor (HIF-1
Malignant tissues often express higher levels of LDH-A compared to non-malignant tissues, which suggests an association between LDH-A and malignant transformation [26]. Inhibition of LDH-A results in stimulation of mitochondrial respiration and compromises the ability of tumor cells to proliferative under hypoxia. Rizwan et al. [27] transfected murine cell models of breast cancer with plasmids to specifically knockdown expression of the LDH-A gene. LDH-A knockdown cells had lower glycolytic rates when compared to that of wild-type cancer cells, which corresponded to an increase in mitochondrial respiration and a lower rate of lactate production both in vivo and in vitro. In vivo, LDH-A depleted tumors had significantly smaller tumor volumes with lower doubling times when compared to wild-type tumors, which corresponded with a reduction and delay in the development of metastases. In a similar study, Fantin et al. [22] used mammary tumor cells with LDH-A knockdown to demonstrate that reduced levels of the enzyme led to reduced cellular transformation and delayed tumor formation. Cells with decreased LDH-A activity had increased oxygen consumption and compromised tumorigenic potential.
Le et al. [28] inhibited LDH-A using either short interfering RNA (siRNA) or a small-molecule inhibitor (FX11 [3-dihydroxy-6-methyl-7-(phenylmethyl)-4-pro pylnaphthalene-1-carboxylic acid]). FX11 is a small molecule that preferentially inhibited LDH-A as opposed to LDH-B. Reduction of LDH-A expression within both human B-lymphoma and pancreatic cancer xenograft models led to ineffective mitochondrial respiration, depleted cellular energy levels, increased oxidative stress, and triggered cell death.
High LDH-A expression is also associated with greater metastatic potential. In a retrospective analysis of patients with advanced small cell lung cancer, elevated serum LDH was correlated with a higher incidence of liver and bone metastases [29], and was an independent predictor of mortality after adjustment was made for age and performance status. Patients with highly elevated serum LDH tended to have more metastatic sites.
When LDH-A was silenced in murine pancreatic cancer cells, the resultant tumors were significantly smaller when compared with wild-type cancer cells [30]. The LDH-A deficient pancreatic cancer cells also secreted less lactate into the surrounding culture. In prostate cancer cells, silencing of LDH-A also resulted in an altered tumor metabolism and microenvironment [31]. The rate of cancer cell apoptosis was significantly increased after LDH-A was inhibited and the number of migrated cells was dramatically decreased in vitro relative to wild-type cancer cells. These preclinical and clinical data strongly suggest that LDH-A is a key enzyme in tumor metabolism, growth and metastatic ability.
Role of LDH-A in resistance of tumor to therapy
By producing lactate and altering the tumor microenvironment, LDH-A can induce resistance to che- motherapy. Maiso and colleagues [32] showed that both HIF-1
In breast cancer cells, those that were resistant to paclitaxel had increased LDH-A expression and activity [33]. When LDH-A was downregulated by siRNA, sensitivity to paclitaxel was increased by at least two-fold in paclitaxel-resistant cells. When paclitaxel-resistant cells were treated with oxamic acid, a specific LDH-A inhibitor, and taxol, these cells displayed a high rate of apoptosis. The therapeutic combination of paclitaxel and oxamic acid was more effective in killing paclitaxel-resistant cells compared to either treatment alone, suggesting a synergistic inhibitory effect. While this study provided promising evidence for using LDH-A inhibition as an area of targeted therapy, further research is required to prove that downregulation of LDH-A mediated sensitization of breast cancer cells to paclitaxel is indeed a consequence of inhibition of glycolysis.
Acidic extracellular microenvironment created by high LDH-A level, generates a drug bioavailability barrier called ion trapping phenomenon, which negatively impacts the efficacy of weak base chemotherapeutic agents. According to the Henderson-Hasselbach equation, for a non-ionized weak base chemotherapy B, which is permeant across the cell membrane, with the corresponding ionized impermeant conjugate acid BH
These observations taken together suggest that LDH-A is more than a marker of tumor burden. Instead, LDH-A seems to have a tumor-protective effect that could relate at least in part to the changes in the tumor microenvironment.
LDH-A promotes immune escape for neoplastic cells
The tumor microenvironment
The tumor microenvironment is a network of extracellular matrix, cells and soluble factors [36, 37, 38]. One of the key soluble factors within the microenvironment is lactate. Excessive levels of lactate produced by LDH-A within tumor cells are excreted from cytoplasm by monocarboxylate transporters (MCT), which catalyze the proton-linked transport of lactate across the plasma membrane. The exported lactate leads to local acidosis of the tumor microenvironment, lowering the extracellular pH to 6.0–6.5 [37]. Lactate has various immune suppressive effects, as described in detail below.
Within the tumor microenvironment, there are im- mune-suppressive cells and immune-stimulatory cells. Cancer-associated immune suppressor cells include tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs). TAMs are pro-inflammatory cells that promote tumor growth via chemokines and cytokines such as interlukin 6 (IL-6), IL-23, IL-12 and tumor necrosis factor alpha (TNF-
Tumor-associated macrophages (TAMs)
TAMs are one of the most common stromal cell types found in tumors. They are enriched in well-perfused areas where they potentiate tumor growth and invasion. Carmona-Fontaine et al. [40] used computational modeling to show that differential sensitivity to acidic microenvironment created by ischemia/hypoxia and lactic acid between cancer cells (specifically aggressive metastatic breast adenocarcinoma cells) and macrophages was sufficient to generate particular spatial localization patterns for TAMs within tumors. TAMs in vivo are enriched in perivascular and cortical areas, where concentration of lactate is less in contrast to the neoplastic cells that can thrive in lactate rich area.
Lactate produced by tumor cells is diffused to the well-perfused areas and picked up by TAMs through MCTs on the cell membrane, where it can induce transcription of vascular endothelial growth factor (VEGF) and arginase 1 (Arg1) (Fig. 1). Lactic acid alone was sufficient to induce VEGF and Arg1 expression in macrophages in a dose-dependent manner [41]. Compared to all other cells within the tumor combined, TAMs express the highest levels of VEGF and Arg1 messenger RNA. Upregulation of VEGF and Arg1 in macrophages induce neovascularization and provide substrates for cancer cell proliferation [42, 43]. Arg1 is also able to induce immune suppression by depleting L-arginine in the microenvironment. Macrophages use L-arginine to synthesize nitric oxide (NO) and polyamines through inducible NO synthase and arginase, respectively [43]. The released NO contributes to the tumoricidal activity of macrophages, whereas polyamines may promote the growth of tumor cells. Depletion of L-arginine inhibits T-cell proliferation and downregulation of CD3
Lactate also polarizes TAMs to differentiate into a so-called “M2” macrophage-like state, favoring tumor growth. M2 macrophages are involved in tissue repair, wound healing and use oxidative metabolism for fuel. This is in contrast to M1 macrophages that act as first line of defense against bacterial infections and obtain energy through glycolysis. Once inside TAMs, lactic acid induces an transcriptional profile that induces differentiation into M2 macrophages [44]. Macrophages stimulated with lactic acid also result in significantly larger tumors than non-lactic acid stimulated cells. Colegio et al. [41] injected mice with murine tumor models of Lewis lung carcinoma (LCC) and melanoma cell lines and compared tumor growth when injected with lactic acid-stimulated macrophages. When exposed to lactic acid-stimulated macrophages, both LCC cells and melanoma cells resulted in larger tumors than with co-injection of control-medium-stimulated macrophages.
Shime et al. [45] showed that tumor-derived lactate is a pro-inflammatory mediator by stimulating IL-23 production from monocytes and macrophages. IL-23 is overexpressed in and around tumor cells, where it induces local inflammation and tumor growth by inducing the production of IL-17 and IL-22. IL-23 also promotes the expression of matrix metalloprotease 9 to increase angiogenesis and metastatic behavior and reduce CD8
Collectively, these findings suggest a model in which TAMs are recruited to well-perfused areas of the tumor microenvironment, where tumor-derived lactic acid induces HIF-1
Myeloid-derived suppressor cells (MDSCs)
MDSCs contribute greatly to tumor evasion from immune attack by controlling the responses of cytotoxic cells such as T cells and NK cells and by migrating into the tumor site and differentiating into highly immune suppressive TAMs. MDSCs regulate and suppress T cells by taking up antigens, processing them and presenting them to CD8
HIF-1
NK cells from LDH-A-depleted tumors had improved cytolytic function against neoplastic cells. Addition of exogenous lactate abrogated these effects to a great extent; lactate promoted the survival and proliferation of MDSCs generated from bone marrow cells via granulocyte macrophage colony stimulating factor (GM-CSF) and IL-6, which interfered with the lytic function of NK cells [30].
Natural killer (NK) cells
The strong link between LDH-A and NK cells has been reported in a few studies. In the same study mentioned before, Husain et al. [30] also used mouse models of pancreatic cancer to demonstrate the functional link between LDH-A and NK activity. Tumor cells with high LDH-A activity could significantly decrease NK cytolytic activity compared to tumor cells without LDH-A. A high lactate concentration also decreased cytotoxic activity of NK cells independent of the acidity of the culture medium when the culture medium was treated with varying concentrations of hydrochloric acid or lactic acid. Lactate-treated NK cells expressed lower levels of perforin and granzyme B when compared to untreated cells. These findings suggest that the observed low cytotoxic activity of NK cells is engendered by lactate, resulting in reduced expression of perforin and granzyme B pathway.
LDH-A also promotes immune escape by inducing expression of certain ligands that allow tumor cells to evade host immune cells. Crane et al. [48] found that sera from patients with glioblastoma multiforme have elevated amounts of LDH, which correlated directly with expression of the natural killer group 2, member D (NKG2D) ligands on circulating host monocytes rather than tumor cells. In fact, glioblastoma-derived LDH5, which is composed only of LDH-A subunits, was sufficient to induce an increase in the amount of NKG2D ligand expression on monocytes. Importantly, when cultured with healthy monocytes, the sera from patients with glioblastoma multiforme was able to induce transcription of NKG2D, suggesting that LDH-A in the plasma was able to induce NKG2D ligand expression on healthy monocytes. Interactions between NKG2D ligand-bearing myeloid cells and NK cells down-modulate NKG2D receptors on NK cells and impair their ability to attack and eliminate tumors. Chronic exposure of NK cells to NKG2D ligand-bearing cells causes down-modulation of the NKG2D receptor on NK cells and inactivation of the NKG2D cytotoxic pathway [49]. For example, freshly isolated tumor-infiltrating NK cells from patients with glioblastoma multiforme had low expression of NKG2D and impaired NKG2D-dependent function [48]. Crane et al. also found that in a small cohort of patients with recurrent glioblastoma multiforme, there was a decrease in the amount of NKG2D ligand expression on circulating monocytes after surgical reduction of the tumor, suggesting that ligand expression is dependent on tumor volume. Therefore, LDH, produced by tumor cells, is correlated with an increased expression of NKG2D ligands on immune cells, allowing the tumor cells evade the host’s immune system. NKG2D ligands have also been found on circulating monocytes isolated from patients with breast, prostate, and hepatitis C virus-induced hepatocellular carcinomas [48], suggesting that tumor-derived LDH-A can induce NKG2D ligands on myeloid cells in these malignancies as well, subverting antitumor immune responses.
T cells
Several studies have shown a link between tumor-derived lactate and T cell function. Fischer et al. [50] established that tumor-derived lactic acid suppressed cytotoxic T-lymphocytes (CTLs) proliferation in a dose-dependent manner. They first demonstrated that serum lactate levels was significantly elevated in patients with high tumor burden. They then incubated cytotoxic T-lymphocytes (CTLs) in a lactate-rich med- ium, which led to cell death. In contrast, treatment of CTLs in an H
In an attempt to find a link between cancer metabo- lism and immune cell infiltration, Singer et al. [51] used renal cell carcinoma cells to show that cancer cells strongly expressed LDH5 and GLUT-1 compared to their nonmalignant counterparts, which is in line with the finding that tumor cells secrete high amounts of lactate. In addition, there was an inverse relationship between GLUT-1 expression and the number of CD8
Dendritic cells
Dendritic cells (DCs), a type of antigen-presenting cells, are important for activation of specific antitumor T-cell responses. Circulating and tumor-associated DCs are phenotypically and functionally defective [52] whereby these tumor-infiltrating DCs have decreased IL-2 production and exist as an immature phenotype. Defective DCs represent one of the mechanisms of tumor evasion from immune system control. Vascular endothelial growth factor (VEGF), produced by virtually all tumor cells, is one factor responsible for the defective function of these cells. VEGF binds to hematopoietic progenitor cells and, via inhibition of transcription factor NF-kB, inhibits their differentiation into mature DCs, thereby leading to tumor-associated DCs that exist in inactive states [14]. Animal models have shown that the type, phenotype and amount of tumor-associated dendritic cells (TADCs) are dynamic over time and influence disease progression at different stages of tumor growth. As tumor burden increases, the number of TADCs have been demonstrated to increase, which was associated with a concomitant loss of T cell infiltration [53]. Tumor cells suppress the function of DCs or alter the tumor microenvironment so that immune-suppressive dendritic cells are recruited.
Lactic acid has been shown to modulate the activation and antigen expression of immune-suppressive DCs. Gottfried et al. [54] used multicellular spheroids of melanoma and prostate carcinoma cell lines that better mimicked the tumor microenvironment to show that lactic acid altered the function of DCs and promoted a tumor-associated DC phenotype that suppresses the host’s immune system. Addition of lactic acid during DC differentiation in vitro induced a phenotype comparable with tumor-infiltrating DCs that were generated within melanoma and prostate carcinoma spheroids. In contrast, hypoxia and acidification alone had little effect on DC differentiation in vitro.
In a spontaneous murine mouse model of aggressive ovarian cancer, tumor growth was associated with increasingly dense immune infiltrates, which included DCs, macrophages, MDSCs and T cells [55]. In the early stages of ovarian cancer progression, depletion of infiltrating DCs led to accelerated tumor growth. As the tumor progressed, the tumor microenvironment caused a phenotypic switch of DCs to TADCs that had immune-suppressive effects. Coinciding with the expansion of immune-suppressive DCs activity in the tumor microenvironment, tumors abrogate protective immunity and start growing in an aggressive manner. Correspondingly, late depletion of TADCs led to inhibition of tumor growth.
Lactate also leads to a concentration-dependent inhibition of T-cell proliferation, by influencing the functional activity of dendritic cells. Lactate suppresses the production of IL-6, IL-12 and TNF-
Monocytes
Tumor-infiltrating monocytes also exhibit protumoral characteristics, including decreased TNF secretion and increased IL-10 or IL-23 production. Peter et al. [56] obtained monocytes from healthy donors to analyze the global effects of lactic acid on gene expression during monocyte activation. Monocytes were cultured with lactic acid, sodium lactate or acidic pH in vitro. Lactic-acid significantly delayed the upregulation of the majority of lipopolysaccharide (LPS)-induced genes, important monocyte effector proteins like cytokines (including TNF and IL-23) and suppressed the genes coding for the chemokines (including CCL2 and CCL7) compared to sodium lactate or acidification. This global alteration of normal monocytes could be one of the mechanisms of how the host’s immune system is altered by tumor-derived metabolites [56].
LDH-C and the testis
Discussion of LDH would not be complete without mentioning LDH-C, which is a sixth isozyme found specifically within mature human testis. LDH-C was the first testicular specific isozyme discovered in male germ cells [57]. The synthesis of LDH-C in the testis takes place during sexual maturation, when spermatids prefer lactate as an energy source over glucose, fructose and pyruvate [58].
During the last two decades of investigation, there have been identification of testis-specific genes, which are expressed only in the germinal epithelium of the testis and not in somatic tissues. Six out of 21 germ cell specific genes have been detected in tumors. The sperm-specific LDH-C gene has been shown to express in a broad spectrum of human tumors, with high frequency in lung cancer, melanoma, and breast cancer [59]. Ectopic activation of testis-specific genes in cancer is a frequent phenomenon. Expression of LDH-C in tumors is not induced by hypoxia nor is it mediated by gene promotor demethylation. As LDH-C has a preference for lactate as a substrate, LDH-C activation in cancer may depend on lactate for cellular energy production [59].
Within the testis, the somatic Sertoli cell presents several common metabolic features analogous to cancer cells, aligning with Otto Warburg observations on metabolism in cancer cells. The somatic Sertoli cells present a high glycolytic flux to ensure the production of high lactate levels and factors required for the development of germ cells. However, Sertoli cells only proliferate during a specific time period and in all species, they cease to divide in adulthood. Therefore, Sertoli cells have a Warburg-like metabolic behavior without the uncontrolled mitotic proliferation seen within cancer cells that is associated with stimulation of growth factors. The high glycolytic flux in Sertoli cells support the nutritional needs of the developing germ cell, but is not associated with cell proliferation [60].
LDH-C has been a unique target for contraception in both males and females and offers potential future for cancer immunotherapy [59].
Concluding thoughts and future directions
In summary, LDH is a key enzyme involved in glycolysis, the preferred metabolic pathway neoplastic cells undergo, resulting in the accumulation and secretion of lactic acid into the extracellular space. Even though the link between glycolysis and cellular proliferation is complex, as suggested by Sertoli cells, there is no denying that LDH could be a marker of rapid cellular metabolism in highly proliferative neoplastic cells. In addition, elevated serum LDH may be a marker for tumor-derived immune suppression; thereby, offering a potential explanation as to why it is a negative prognostic biomarker for a variety of hematologic and solid neoplasms. By promoting immune suppression in the tumor microenvironment, LDH-A is able to stimulate tumor growth, metastasis and resistance to chemo/radio/targeted therapy.
To date, research has focused on the role of LDH-A within tumor cells and tissues. Some of the more promising findings have been the effect of specific LDH-A inhibitors, such as oxamic acid and FX11, on reversing therapy resistance. Investigation into the role of specific LDH isoenzymes, both LDH-A and LDH-C, within malignant cells and tissues may open an exciting area of targeted therapy. Moreover, with the discovery of modern and effective immunotherapies for different malignancies in the past few years, elevated LDH can be used in designing clinical trials for appropriate patient selection, and perhaps decreasing the cost by predicting response.