
Abstract
Brain plasticity and metabolism are tightly connected by a constant influx of peripheral glucose to the central nervous system in order to meet the high metabolic demands imposed by neuronal activity. Metabolic disturbances highly affect neuronal plasticity, which underlies the prevalent comorbidity between metabolic disorders, cognitive impairment, and mood dysfunction. Effective pro-cognitive and neuropsychiatric interventions, therefore, should consider the metabolic aspect of brain plasticity to achieve high effectiveness. The adipocyte-secreted hormone, adiponectin, is a metabolic regulator that crosses the blood-brain barrier and modulates neuronal activity in several brain regions, where it exerts neurotrophic and neuroprotective properties. Moreover, adiponectin has been shown to improve neuronal metabolism in different animal models, including obesity, diabetes, and Alzheimer’s disease. Here, we aim at linking the adiponectin’s neurotrophic and neuroprotective properties with its main role as a metabolic regulator and to summarize the possible mechanisms of action on improving brain plasticity via its role in regulating the intracellular energetic activity. Such properties suggest adiponectin signaling as a potential target to counteract the central metabolic disturbances and impaired neuronal plasticity underlying many neuropsychiatric disorders.
Impaired brain plasticity is a hallmark of cognitive- and mood-related disorders, such as Alzheimer’s disease (AD) [1] and depression [2], which is emphasized by the high comorbidity among such conditions [3]. Brain function is tightly connected with the peripheral metabolism given the brain’s need for a continuous influx of peripheral glucose to support its high metabolic demands [4], such that metabolic dysfunctions eventually result in disruption of brain activity and impaired neuronal plasticity [4]. Not surprisingly, metabolic disorders are commonly associated with cognitive and mood impairments [5, 6], whereas AD itself is currently characterized by some as type 3 diabetes [7]. Given that metabolic disorders are an ever-growing problem [8, 9], it is expected that the incidence of cognitive- and mood-related problems will increase in the coming years. Such entangled nature of metabolism and brain plasticity suggests that effective treatments must tackle both ends: metabolism and plasticity, at the same time [5].
Adiponectin is a cytokine with remarkable pleiotropic properties. It has been shown to be anti-inflammatory, anti-atherosclerotic, anticarcinogenic, neuroprotective, pro-cognitive, and antidepressant [10, 11]. Being a cytokine with receptors expressed across several physiological systems [10], including the brain [12], adiponectin functions as a major metabolic regulator. Its relevance for proper brain functioning has been demonstrated by marked neurophysiological and behavioral impairments displayed by adiponectin deficient mice [13], resembling an AD-like pathology [14]. On the other hand, increasing central adiponectin signaling has been remarkably effective in counteracting central metabolic dysfunctions and increasing brain plasticity [15– 17].
Although genetic factors have an impact on metabolic functions, lifestyle factors also play a critical role [18]. In this regard, chronic psychological stress is a significant risk factor for developing metabolic dysfunctions [4]. Increasing energy availability is an elementary allostatic response during any stress reaction [4, 19]. However, it poses a challenge to the individual’s metabolic system if such stress is prolonged over days, weeks, or longer [4, 19]. Likewise, chronic stress is linked to central metabolic dysfunction that is followed by impaired neuroplasticity [20, 21]. Although chronic stress-associated neuronal atrophy is usually associated with downregulation of neurotrophic support [22, 23], the mechanisms by which stress specifically reduces neurotrophic support are largely unknown. There is a resurging interest in the metabolic changes induced by stress and how they could underlie stress-induced neuronal impairments [5, 24]. As remembered by Picard and colleagues, glucocorticoids were originally termed after their influence over glucose metabolism [24].
In this review, we aim to link the adiponectin’s neurotrophic and neuroprotective properties with its main role as a metabolic regulator, suggesting the improvements in brain plasticity observed by adiponectin treatment result from the regulation of the intracellular energetic activity. We first review the adiponectin signaling as a regulator of the peripheral and central metabolism and its relevance for brain plasticity, followed by a discussion on the negative impact of chronic psychological stress on neurodegeneration and the central metabolism, whereas cognitive and mood impairments are mentioned as representative behavioral changes associated with plasticity deficits. Finally, adiponectin’s neurotrophic and neuroprotective properties are reviewed and considered along with the restored intracellular metabolic functions.
CENTRAL ADIPONECTIN SIGNALING REGULATES CELLULAR METABOLISM AND SUPPORTS NEURONAL PLASTICITY
Adiponectin regulates peripheral metabolism
Adiponectin synthesis is controlled by a multitude of transcriptional factors, including the peroxisome proliferator-activated receptor (PPAR) gamma, and undergoes several post-transcriptional and post-translational steps involving different chaperones before its secretion [25]. Adiponectin synthesis and secretion are inversely proportional to the visceral adipose depot [26] and the adipocyte size [27]. Although the mechanisms for such negative modulation are largely unknown, the tumor necrosis factor (TNF)-α has been shown to negatively modulate circulating adiponectin levels as treating mice with recombinant TNF-α reduces circulating adiponectin levels [28] and pharmacologically blocking TNF-α in subjects with metabolic syndrome increases it [29]. Structurally, the 244-aminoacid protein consists of a globular domain, a collagen-like domain, a species-specific domain, and a signal peptide (e.g. full-length form) [10, 11]. The full-length adiponectin undergoes oligomerization and circulates as trimer, hexamer, or high-molecular-weight multimers, whereas trimers can be cleaved to globular adiponectin (Fig. 1) [11, 30].
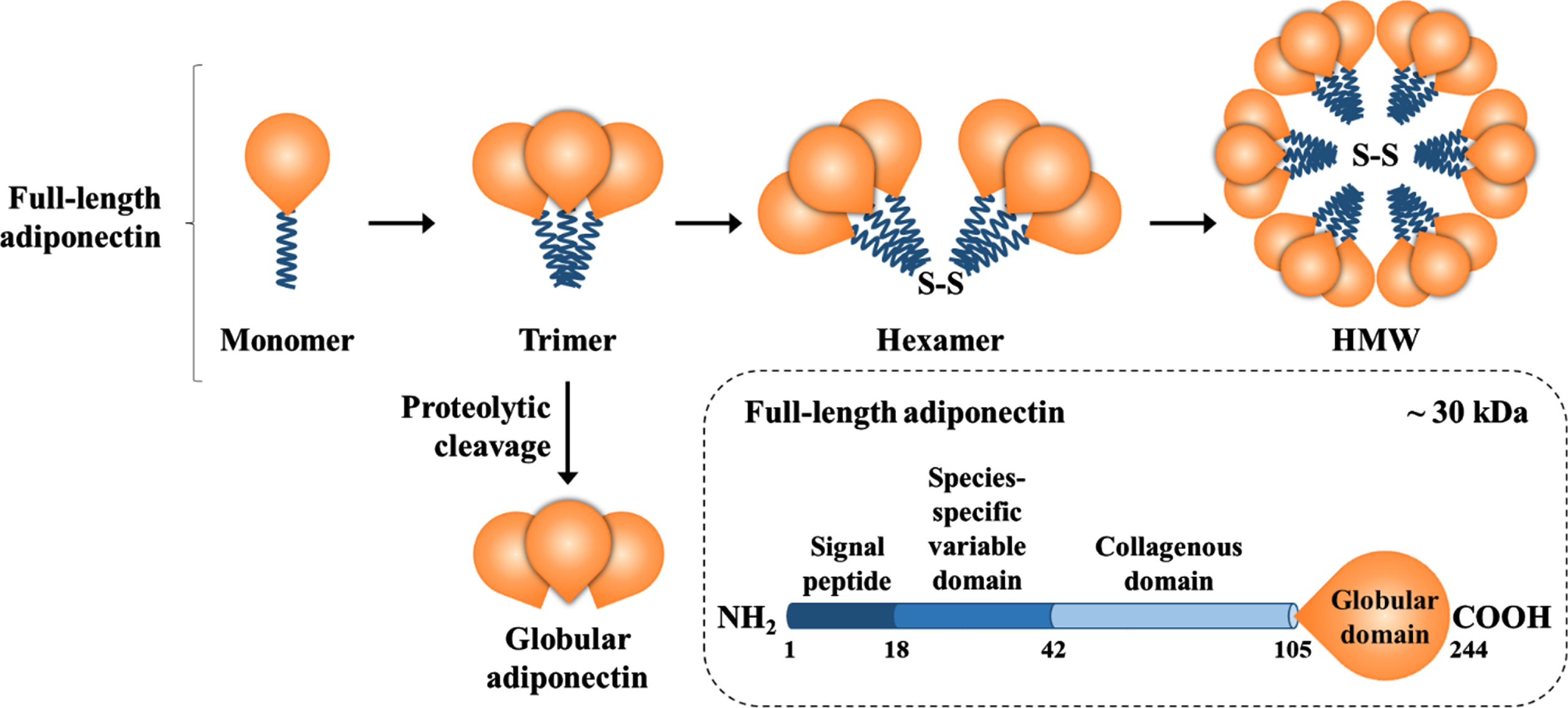
Adiponectin composition and isoforms. Full-length adiponectin is composed of its three main domains plus a globular domain, which can be cleaved and circulate as globular adiponectin. Full-length adiponectin undergoes oligomerization and circulates as a trimer, hexamer, or high-molecular-weight multimer, whereas trimers and hexamers can cross the blood-brain barrier.
Adiponectin exerts its physiological functions mainly through the activation of two specific receptors, namely the AdipoR1 and AdipoR2 [31]. Whereas AdipoR2 has a moderate affinity for both adiponectin isoforms, AdipoR1 has a higher affinity for globular adiponectin in comparison with the full-length isoform [31]. Both receptors interact with the adaptor protein containing a pleckstrin homology domain, a phosphotyrosine domain, and a leucine zipper motif (APPL1) as their key mediator coupling receptor activation with downstream signaling pathways [32]. As an endosome-associated adaptor protein, APPL1 can modulate signaling specificity and crosstalk by anchoring effectors of different signaling pathways [33, 34]. APPL1 promotes the crosstalk between adiponectin receptors and its downstream signaling targets, including the activation of the PPARα [32, 35], the AMP-activated protein kinase (AMPK) [35], and other proteins associated with the insulin signaling pathway, such as phosphatidylinositol 3-kinase (PI3K) and protein kinase B (Akt) [36] (Fig. 2A).
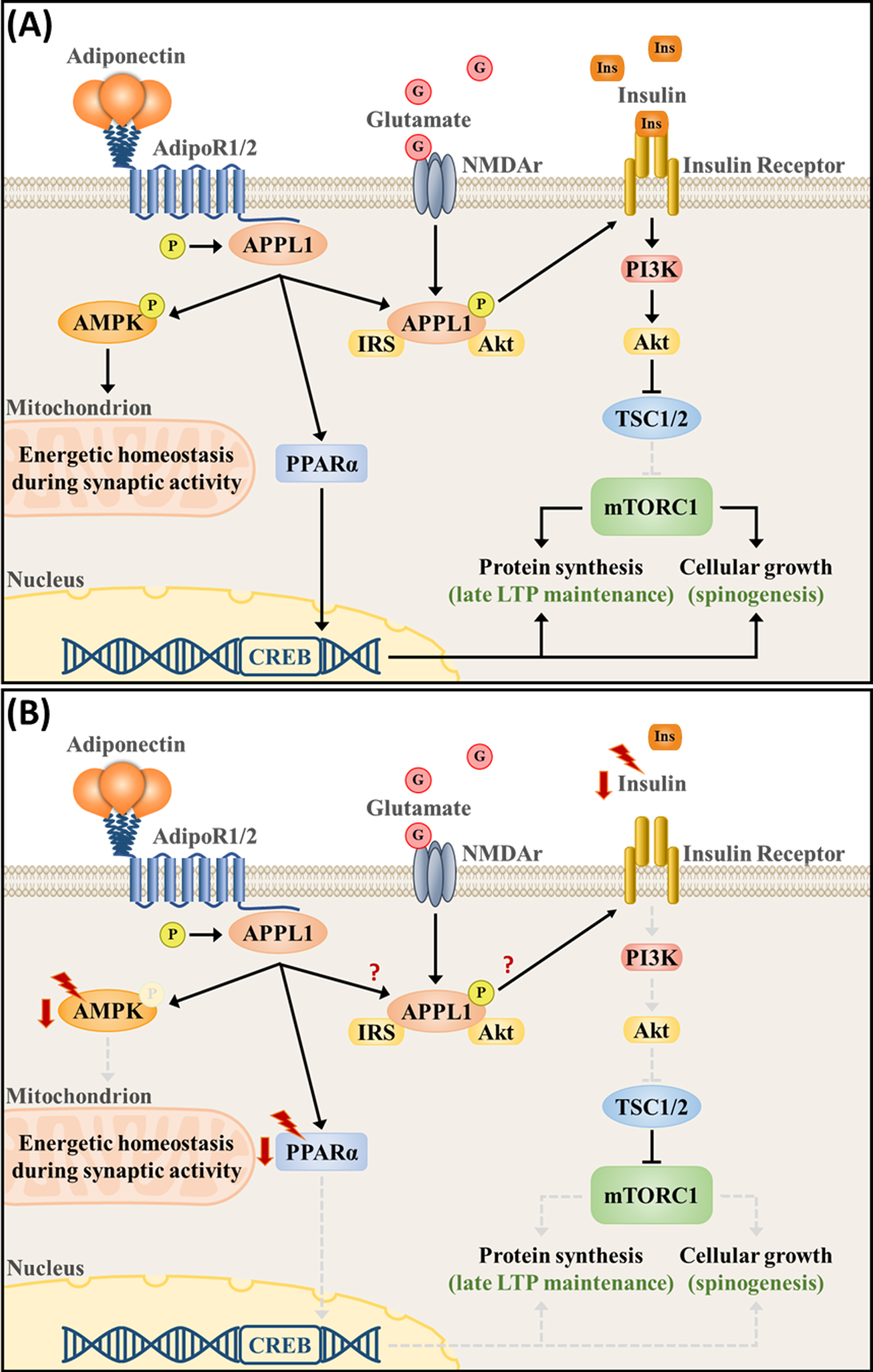
Adiponectin signaling pathway regulates intracellular metabolism and neuronal plasticity in (A) physiological- and (B) chronic stress-associated conditions. (A) Activation of the adiponectin receptors, which are present at synapses, orchestrates signaling pathways that are essential for proper bioenergetic balance and plasticity. AdipoR1/2 phosphorylates the APPL1 and activates the AMPK and PPARα pathways, whereas APPL1 itself forms a complex with NMDA receptors and couples neuronal activity with the activation of the insulin signaling pathway (PI3K/Akt). PI3K/Akt induces mTORC1 activity, protein synthesis, and cellular growth by suppressing TSC1/2. Upon synaptic activation, increased AMPK signaling is necessary to upregulate substrate oxidation, mitochondrial respiration and biogenesis, maintaining the intracellular energetic reserves necessary for LTP maintenance. PPARα, on the other hand, directly upregulated CREB transcription. (B) Chronic stress downregulates PPARα and AMPK expression and phosphorylation, as well as downregulates the expression of upstream modulators of the PI3K/Akt pathway, such as vascular endothelial growth factor and insulin. Currently, a direct connection between APPL1 and stress-associated deficits remains unknown.
PPARα is a transcription factor that functions as a lipid sensor. PPARα responds to starvation by regulating the expression of several genes associated with fatty acid metabolism, including fatty acid uptake and oxidation [37]. In parallel, PPARα also downregulates several proinflammatory factors [37]. AMPK, on the other hand, functions as the cellular energy sensor. AMPK is activated upon circumstances that pose a challenge for adenosine triphosphate (ATP) production, such as hypoxia and glucose deprivation [38– 40]. Being the canonical catabolic pathway, AMPK senses an increase in adenosine monophosphate to ATP ratio, leading to the phosphorylation of proteins that regulate the catabolic process, such as increased gluconeogenesis and fatty acid oxidation, while suppressing anabolic mechanisms, such as glycolysis and protein synthesis, resulting in increased energy availability [38].
APPL1 also mediates the crosstalk between the adiponectin and insulin signaling pathways, as suggested by the insulin resistance and impaired insulin signaling observed in APPL1 knockout mice [36]. Specifically, APPL1 facilitates the interaction of insulin-receptor substrates 1 and 2 with the insulin receptor β, which is dependent on the APPL1 phosphorylation on the Ser401 site [36]. Interestingly, APPL1Ser401 phosphorylation is also promoted by adiponectin, which mediates the adiponectin’s insulin-sensitizing properties [36]. However, insulin is necessary for the downstream activation of the PI3K/Akt cascade [36]. Such pathway is part of the canonical anabolic pathway as it inhibits the tuberous sclerosis proteins 1 and 2 activity (which suppress the activity of the mammalian target of rapamycin complex 1 [mTORC1]) and activates the mTORC1, resulting in protein synthesis and cell growth [38, 41] (Fig. 2A).
On the one hand, adiponectin signals the initiation of catabolic reactions through activation of the AMPK pathway, interrupting protein synthesis and cell growth. On the other hand, it facilitates anabolic processes through the PI3K/Akt pathway, fostering protein synthesis and cell growth. It is, therefore, intriguing how adiponectin activates antagonistic processes simultaneously. One possible explanation relies on the organism’s nutritional state. In the absence of glucose and, hence, insulin (starvation), the adiponectin-induced activation of AMPK and PPARα pathways prevails, resulting in the AMPK activation of the tuberous sclerosis proteins 1 and 2 and consequent suppression of mTORC1 (catabolism) [38, 41]. On the other hand, in the presence of insulin and growth factor stimulation (nutrient-rich state), adiponectin facilitates the PI3K/Akt pathway activation, which interacts with the glycogen synthase kinase 3 beta to inhibit AMPK activity [42], interrupting catabolic processes and leading to protein synthesis and cell growth through activation of the mTORC1 (anabolism). Activation of such downstream signaling pathways evidences the adiponectin’s role as an insulin sensitizer to improve glucose metabolism and fatty acid oxidation, eventually counteracting hyperglycemia in metabolic disorders [43– 46].
The APPL1, PPARα, AMPK, and PI3K/Akt are also expressed in the neuronal tissues, where they have an important role in supporting neuronal metabolism and plasticity.
Adiponectin downstream signaling pathways regulate neuronal metabolism and plasticity
Brain activity relies on a disproportional amount of energy requirements compared with the rest of the body. Roughly 20% of the body’s metabolic resources are used by the central nervous system (CNS), although it accounts for only 2% of the total body weight [47]. Since the brain contains virtually no energetic reserves, central glucose levels are maintained through a constant influx of peripheral glucose through the blood-brain barrier, which is facilitated by the glucose transporter 1 [4]. Such high energetic requirements result mostly from synaptic activity (i.e., generation of action potentials as well as re-establishment and maintenance of membrane potential) [48, 49]. Therefore, excitatory glutamatergic transmission largely determines the brain’s energetic demands [48, 49]. Not surprisingly, neuronal mitochondria are mostly located at synapses where oxidative respiration is tightly connected with neuronal activation [49]. Given that energy requirements are directly proportional to synaptic activity, increased α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor trafficking due to synaptic potentiation naturally results in increased energy demands [49]. Therefore, tight regulation of intracellular metabolism is the basis for proper synaptic and structural plasticity.
PPARα is expressed in all hippocampal subfields [50] and has been implicated in glutamate metabolism and transmission [51]. It can promote glutamate transporter 1 endocytosis in astrocytes and downregulate the level of endogenous glutamate receptors antagonists, such as kynurenic acid [51]. In the hippocampus, it indirectly modulates the expression of synaptic-related genes (e.g., GluN2A and GluR1) via direct upregulation of cAMP response element-binding protein (CREB) transcription, which is necessary for adequate calcium influx and acquisition of long-term spatial memory [50] (Fig. 2A).
In the hypothalamus, the AMPK signaling pathway serves as a master metabolic sensor. The different hormones associated with fasting (e.g., ghrelin) or feeding (e.g., leptin) activate or inhibit hypothalamic AMPK, respectively, and determine the subsequent metabolic functions based on AMPK activity (e.g., decreased energy expenditure and increased food intake) [52]. At the cellular level, constitutive AMPK is not necessary for proper neuronal development, although in vitro overactivation of AMPK using its agonist, 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR), impairs axogenesis by inhibiting mTORC1 activity [53]. Nutrient sufficiency is required for the brain-derived neurotrophic factor (BDNF)-induced activation of mTORC1 [54]. Upon glucose starvation, increased AMPK activity directly blocks BDNF-induced activation of mTORC1 and subsequent protein synthesis [54]. Furthermore, AMPK is recruited upon synaptic activation to sustain intracellular ATP levels, upregulating mitochondrial respiration and glycolysis to meet the energy requirements upon cellular activation [55]. Energy-depleted cells fail to activate the mitogen-activated protein kinase pathways [55]. Blocking AMPK activity impairs long-term potentiation (LTP) and inhibits the protein-synthesis-dependent late phase of LTP in the hippocampal cornu Ammonis 1 (CA1) subregion, which leads to deficits in contextual fear memory learning [55]. Moreover, AMPK is also necessary for activity-induced immediate early-genes expression (Arc, Egr1, cFos) [55]. AMPK, therefore, signals cellular metabolic adjustments upon synaptic activation (Fig. 2A).
Insulin receptors are also present at the synapse level [56], where insulin-receptor substrates are translocated upon increased N-methyl-D-aspartate (NMDA) receptor activity [57]. The insulin signaling in dendritic spines is relevant for the maintenance of activity-dependent synaptic efficiency and experience-induced structural plasticity in terms of dendritic complexity [58]. The NMDA receptor forms a complex composed of 77 proteins, amidst which APPL1 can be found [59]. APPL1 directly interacts with the NMDA receptor complex and couple NMDA receptor activity with recruitment and activation of the PI3K/Akt pathway in postsynaptic densities [60] (Fig. 2A). Such APPL1-mediated interaction with the PI3K/Akt pathway is necessary for appropriate spine and synapse formation, such that knocking down APPL1 activity in cultured hippocampal neurons impairs dendritic spine formation [61]. In parallel, dendritic spine-located APPL1 senses synaptic activity and is translocated to the nucleus upon neuronal activation, where it facilitates transcription necessary for late-phase LTP maintenance. Blocking APPL1 activity impairs late LTP but not LTP induction nor early LTP [62].
Whether adiponectin can be expressed in the nervous tissue is still debatable. However, low-molecular-weight adiponectin, both the globular and full-length isoforms, can cross the blood-brain barrier [63, 64]. Moreover, adiponectin receptors are found in synapses [13] and are widely expressed throughout the CNS, including the ventral tegmental area [65], dorsal raphe nucleus [66], medial prefrontal cortex (mPFC), hippocampus, and amygdala [12]. Given the relevance of the adiponectin’s downstream targets for the modulation of central metabolism and maintenance of neuronal activity, adiponectin is likely to be one of the key peripheral factors modulating neuronal metabolism and plasticity [30, 67]. Such rationale has been supported by the impairments observed in mice with chronic adiponectin deficiency [13, 14, 17].
Adiponectin deficiency leads to impaired neuronal metabolism and plasticity
Adiponectin deficiency greatly impairs intracellular metabolism in the CNS. Chronic adiponectin deficiency in aged adiponectin knockout mice leads to hippocampal insulin resistance, which is associated with reduced phosphorylation levels of AMPK, Akt, and PI3K [13, 14, 17], despite normal insulin plasma levels and normal hippocampal insulin receptor expression [14]. Moreover, Akt phosphorylation is impaired in response to intra-hippocampal insulin administration in adiponectin knockout mice [14]. Remarkably, such impairment is reversed by adiponectin in cultured insulin-resistant neuronal cells, since adiponectin pretreatment improves Akt phosphorylation in response to insulin challenge [14].
Disrupted CNS metabolism in adiponectin deficient mice is associated with impaired functional and structural plasticity. Adiponectin knockout mice have reduced presynaptic and postsynaptic protein expression, including synaptophysin (presynaptic), synaptosomal-associated protein 23, synapse-associated protein 102, and postsynaptic density protein 95 (PSD-95) (postsynaptic) [14, 17]. These postsynaptic proteins are associated with NMDA receptors and glutamatergic transmission. In agreement with that, such animals also present reduced levels of the glutamate receptor subunits GluA1, GluN1, and GluN2A (but not GluN2B) along with reduced phosphorylated CREB [13]. Such impairments in glutamatergic transmission are accompanied by reduced synaptic plasticity, dendritic complexity, spine density, and neurogenesis in the hippocampal region [13, 17, 68]. Not surprisingly, chronic adiponectin deficiency leads to impaired spatial learning and memory, contextual and cued fear conditioning, and increased anxiety-like behavior [13, 14, 17]. Moreover, adiponectin knockout mice do not benefit from the physical exercise-induced increase in hippocampal neurogenesis and antidepressant effects [69].
Lifestyle factors greatly interfere with metabolism, circulating adiponectin levels, cognition, and mood. Imbalance in the input/output energy levels due to excessive energy intake and low physical activity levels can lead to increased adiposity, obesity, and diabetes, all associated with reduced circulating adiponectin levels [70– 72], cognitive impairments [74– 76], and reduced structural and functional plasticity [74– 76]. Likewise, chronic psychological stress, which will be thoroughly discussed next, is linked to reduced adiponectin signaling and impaired peripheral and central metabolism.
CHRONIC STRESS IMPAIRS ADIPONECTIN SIGNALING AND DISRUPTS NEURONAL PLASTICITY
Chronic stress impairs peripheral and central metabolism
Stress is an adaptive response to external and internal stimuli, which aims to adjust the physiological functions to cope with the encountered challenges [79]. In this regard, the stress response can arise from metabolic disturbances, such as those induced by dietary and exercise habits (i.e. metabolic stress) [80], or environmental factors, which are perceived as threatening the subject’s well-being (i.e. psychological stress) [81].
In psychological stress, the central survival circuitry detects innate and learned threats and activates the hypothalamus-pituitary-adrenal axis, leading to the release of catecholamines (autonomic response) and glucocorticoids (e.g. cortisol, endocrine response) [82]. Glucocorticoids are key modulators of glucose metabolism [4, 19]. They increase plasma glucose levels and promote gluconeogenesis and glycogen storage in the liver, increase lipolysis and visceral fat, reduce glucose uptake in the skeletal muscles, and antagonize insulin production [4, 19]. Such metabolic adjustments induced by the survival response are an important adaptive response to acute stimuli perceived as threatening. Nonetheless, chronic psychological stress eventually results in hyperglycemia and insulin resistance [4, 19]. Not surprisingly, it is a risk factor for type 2 diabetes and impacts the disease’s prognosis [83]. Here, we focus on the metabolic disturbances associated with chronic psychological stress, henceforth simply referred to as stress.
Psychological stress can be experimentally induced in animals through exposure to naturalistic stressors, such as food, water, or sleep deprivation, or social stressors, such as social isolation and social defeat by exposure to a stronger male aggressor (referred to [84] for in-depth review). Interestingly, it has also been suggested that stress can potentially downregulate adiponectin expression. Dexamethasone, a glucocorticoid receptor (GR) agonist, reduces circulating adiponectin levels and leads to metabolic disorders in rodents [85, 86]. In humans, reduced plasma adiponectin levels were observed in firefighters diagnosed with post-traumatic stress disorder [87]. Moreover, glucocorticoids were shown to reduce adiponectin transendothelial movement, suggesting that stress can reduce adiponectin availability to targeted tissues even if serum concentrations are unchanged [88].
The CNS’s reliance on glucose as its main energetic source renders it vulnerable to such metabolic disturbances. In rodents, chronic stress induces hippocampal hyperglycemia [21], increases hippocampal glycogen synthesis [89], and reduces hippocampal and mPFC mitochondrial respiratory chain activity [90, 91]. Such stress-induced disturbances in the central metabolism were accompanied by increased depression-like [90, 91] and impaired spatial memory [21, 89]. Likewise, intra-hippocampal glucose infusion impairs spatial memory [21]. On the other hand, lowering blood glucose levels pharmacologically rescues spatial memory performance in hyperglycemic stressed mice [21].
The stress-induced impairments on neuronal metabolism and plasticity are largely mediated by decreased PPARα, AMPK, and PI3K/Akt expression and activity.
Chronic stress impairs the adiponectin’s downstream signaling pathways and disrupts neuronal plasticity
In rodents, three isoforms of PPAR are co-expressed in the brain circuits responsible for regulating stress responses, playing a role in several neuropsychiatric disorders by modulating anti-inflammatory and metabolic processes [92]. PPAR-α is one of the three isoforms of the nuclear PPAR family [93], whose physiological functions are associated with cell differentiation, metabolism, and energy homeostasis [94]. Upon binding to endogenous and synthetic ligands, PPARα is transferred to the nucleus to dimerize with the retinoic acid x receptor and bind to the peroxisome proliferative response element to regulate the transcription of the target gene [95]. It is abundantly expressed in the hippocampus, amygdala, prefrontal cortex, thalamic nucleus, vomeronasal nucleus, and ventral tegmental region [96], where it modulates the production of BDNF [97], a critical neurotrophic factor governing neuronal plasticity. Of note, hippocampal PPARα expression is downregulated by chronic stress (Fig. 2B, which leads to increased expression of depression-like behaviors [98].
Chronic stress has also been linked with reduced AMPK activity and expression in several experimental models (Fig. 2B). In vitro, corticosterone significantly reduces AMPK phosphorylation in a time-dependent fashion in both astrocytes (from 2-h to 72-h incubation time) [99] and PC12 cells (24-h incubation time) [100]. In vivo, different chronic stress protocols were shown to reduce AMPK phosphorylation in the mPFC [99], hippocampus [101– 103] and cortex [104]. Noteworthy, the hippocampus and mPFC are two regions that have been classically involved in mood and cognitive performance. Congruently, chronic stress-induced reduction in AMPK signaling was associated with increased depression- and anxiety-like behavior [99, 103] and impaired spatial memory [101– 103].
There are different mechanisms through which decreased AMPK activity can be linked with chronic stress outcomes. First, as previously discussed, AMPK functions as a metabolic switch that transiently activates catabolic pathways to adjust the increased intracellular metabolic demands upon neuronal activation, a process that is essential for further activation of the mitogen-activated protein kinase pathways, protein synthesis, and synaptic plasticity [55]. Second, stress-induced reduction in the central expression of GR results in glucocorticoid resistance and hypercortisolemia [82], which is associated with impaired plasticity and neuronal atrophy [105, 106]. Remarkably, pharmacological activation of AMPK in cultured astrocytes protects against the dexamethasone-induced reduction in GR, whereas its pharmacological or genetic inhibition results in reduced GR expression [99]. Finally, AMPK also upregulates BDNF signaling. One-hour incubation of cultured primary hippocampal neurons with AICAR significantly increases AMPK phosphorylation and BDNF levels in a dose-dependent manner [101]. Likewise, both acute (30 min) [107] and sub-chronic (7 days) [101] systemic administration of AICAR increases hippocampal BDNF levels, which can last for up to 14 days after the last sub-chronic dose administration [101].
Chronic stress can impair the PI3K/Akt signaling pathway by downregulating the expression of upstream modulators, such as the vascular endothelial growth factor [108] and insulin [109] (Fig. 2B). Chronic corticosterone treatment dysregulates the vascular endothelial growth factor expression, resulting in reduced activation of the PI3K/Akt/mTOR signaling pathway [110]. Moreover, chronic stress disrupts the insulin-mediated activation of this pathway, resulting in cognitive impairment in healthy [109], AD [111], and diabetic [112] rodents. The PI3K/Akt pathway is the canonical anabolic pathway, further targeting proteins like the glycogen synthase kinase 3 beta [113], mTOR [114], and forkhead box protein O (FOXO) [115], all involved in protein synthesis and cellular growth. Disrupting these pathways result in an overall shift in the molecular and cellular mechanisms mediating synaptic and structural plasticity.
Finally, the impact of chronic stress on APPL1 expression and phosphorylation, to our knowledge, remains to be explored. However, considering the previously reported relevance of APPL1 in linking glutamatergic activity with the activation of the insulin signaling pathway [60, 61, 116, 117], it is reasonable to speculate that disturbed APPL1 activity likely mediates the deleterious effect of chronic stress on neuronal metabolism and plasticity. Although the specific involvement of APPL1 with mood and cognition is still lacking, APPL1 was found differentially expressed in postmortem brain samples of AD subjects [118]. Specifically, APPL1 was observed to aggregate in granules around hippocampal pyramidal cells, mostly in the CA1 and subiculum areas, a feature present only in AD patients but not in normally aged controls [118]. However, at the current state of the literature, it is not possible to draw a direct connection between APPL1 and stress-associated deficits nor is it possible to indicate it as a therapeutic target.
Targeting the adiponectin’s downstream signaling pathways results in therapeutic response
The hippocampal PPARα has been previously established as a novel antidepressant target [119] given its upregulating properties over the BDNF/CREB signaling pathway [97], which is necessary for the antidepressant effects. Specifically, genetic overexpression of hippocampal PPARα produces antidepressant effects by promoting CREB-mediated BDNF biosynthesis [98]. On the other hand, PPARα-associated antidepressant effects were also linked with the activation of the mesolimbic dopaminergic system [120]. Fenofibrate enriched-diet, a PPARα agonist, can prevent and alleviate chronic stress-induced depression-like behaviors by increasing activity of the ventral tegmental area [120].
As previously addressed, AMPK upregulates BDNF signaling, which is a key mediator of antidepressant treatments like ketamine [121] and physical exercise [122]. In agreement with that, both acute ketamine administration [107] and chronic physical exercise [101, 102] increased hippocampal AMPK activity, whereas pre-treatment of animals with Compound C, an AMPK inhibitor, reduced the ketamine rapid-antidepressant effects [107]. On the other hand, AMPK was also shown to be a key mediator of the physical exercise-associated increase in neuroplasticity and cognition. In stressed mice, six injections with Compound C once every 4 days during 21 days of treadmill running significantly reduced the exercise improved spatial memory and blocked the exercise-associated increase in BDNF, synaptophysin, and PSD-95 mRNA expression [101]. Such blunted effect on synaptic protein expression and neurotrophic support abolished exercise-induced adult neurogenesis, dendritic complexity, and spine density [101, 102].
Directly targeting the AMPK activity, on the other hand, elicits therapeutic effects. In cultured primary hippocampal neurons, AICAR increases dendritic branching and length, which is protective against the corticosterone-induced reduction in dendritic complexity [102]. Moreover, AICAR treatment reduces hippocampal oxidative stress and prevents chronic stress-induced depression, anxiety, and cognitive impairments [103]. Similarly, 14 days of metformin administration, an antidiabetic drug relying on AMPK activation, improved chronic corticosterone-induced depression-like behavior, which was associated with increased expression of GR in the mPFC [99]. Likewise, sub-chronic treatment with AICAR (7 days) improves the cognitive performance of non-stressed mice [101].
PI3K/Akt downstream targets are key mediators of rapid-acting antidepressant drugs, such as the mTORC1-dependent antidepressant effects induced by ketamine [123]. Moreover, FOXO3a, another target of the PI3K/Akt pathway, plays an essential role in depression development and treatment [124]. Studies have shown that mice with FOXO3a gene deletion exhibit depression-like behavior [115], whereas antidepressant drugs, such as venlafaxine [125], exert antidepressant effects by modulating FOXO3a.
In summary, stress-induced downregulation of adiponectin expression and consequent metabolic dysregulation leads to impairments in neuronal plasticity that are largely mediated by a reduction in the activity of the adiponectin downstream signaling pathways. Moreover, directly targeting such pathways significantly restores neuronal metabolism and plasticity, suggesting the increase in adiponectin signaling as a prominent strategy for restoring neuronal metabolism and improving brain plasticity (Fig. 2).
INCREASING ADIPONECTIN SIGNALING AS A THERAPEUTIC STRATEGY FOR IMPROVING CENTRAL METABOLISM AND NEURONAL PLASTICITY IMPAIRMENTS
Adiponectin is the most abundant plasma protein [126], rendering its use as a direct treatment unsuitable. Nonetheless, two different compounds, AdipoRon and Osmotin, have been showing promising therapeutic properties by targeting the adiponectin signaling system. AdipoRon is a synthetic, orally active small molecule that agonizes both adiponectin receptors [127], whereas Osmotin is a plant-based protein homologous with adiponectin [128]. Both compounds have been experimentally tested to counteract neurological impairments associated with central metabolic dysfunctions, including those associated with AD, diabetes, obesity, and others.
Alzheimer’s disease has been increasingly understood as a metabolic disorder, with some classifying it as type 3 diabetes [7, 129]. [17] On the other hand, exogenous administration of adiponectin in several animal models of AD resulted in marked therapeutic effects. The treatment of acute hippocampal slices with adiponectin [130] increased synaptic plasticity in a transgenic animal model of AD, whereas 48 h of in vitro AdipoRon incubation rescued the amyloid β-induced impairments in dendritic complexity and neural stem cells’ proliferation [16]. AdipoRon administered centrally (intracerebroventricular infusion) [16] or orally [15] improved hippocampal insulin sensitivity and neuroinflammation [15], increased dendritic complexity, spine density [15], and cell proliferation [16] in transgenic animal models of AD. Such effects were accompanied by reduced anxiety-like behavior and improved spatial memory [15, 16]. Moreover, in a recent study investigating the therapeutic effects of long-term Osmotin treatment, the authors observed increased hippocampal synaptic protein expression, increased dendritic complexity and spine density, and increased synaptic plasticity in three different models of AD [17].
Interestingly, the impairments in neuroplasticity, cognitive performance, and anxiety-like behavior presented by aged adiponectin knockout mice (mentioned in the previous sections) are associated with increased amyloid β and tau phosphorylation in the hippocampus and frontal cortex [14]. Moreover, adiponectin deficiency further worsens the anxiety and spatial memory impairments in transgenic AD animal models [15]. Therefore, the neurophysiological and behavioral alterations induced by chronic adiponectin deficiency are associated with features resembling an AD-like pathology.
Although targeting the central adiponectin signaling system to counteract AD neuropathology has been promising in animals, the clinical observations regarding adiponectin and cognitive function are rather contradictory [131, 132]. High circulating adiponectin levels are associated with mild cognitive impairment and AD [131, 133], which was also associated with smaller hippocampal volume and increased Aβ deposition in women [134]. Moreover, in a long-term prospective study adiponectin was the only independent factor positively associated with increased risk for developing all-cause dementia and AD during the 13-year follow-up [132]. The mechanisms involved with such adiponectin paradox, which extend beyond AD [135, 136], are still unknown. Adiponectin resistance resulting from reduced expression of adiponectin receptors and/or the downstream adiponectin signaling targets has been suggested as one of the possible mechanisms [136]. Indeed, postmortem samples from patients with AD showed lower expression levels of AdipoR1 in the cortex and hippocampus compared with aged healthy subjects [17].
Obesity and diabetes were the first metabolic disorders to be associated with lower circulating adiponectin levels [44, 137] and to hint at the adiponectin’s role as a metabolic regulator [44, 126]. Streptozotocin-induced diabetes reduces the hippocampal levels of adiponectin [138] whereas high-fat diet-induced obesity reduces the adiponectin receptors’ expression in the hippocampus [139]. Not surprisingly, this is associated with impaired hippocampal functional and structural plasticity, including reduced expression of PSD-95, LTP, dendritic complexity, spine density, and neurogenesis [77, 78, 138]. Physical exercise is a potent treatment for metabolic disorders. Our group has demonstrated that adiponectin is a key mediator of the voluntary wheel running neurogenic and antidepressant effects, both in physiological conditions [69] as well as in diabetic mice [138]. In humans, high-intensity exercise appears to have the strongest effect over peripheral adiponectin expression [140, 141]. High-intensity aerobic exercise increased the high-molecular-weight to total adiponectin ratio in obese men [140], whereas in obese young female adolescents high-intensity interval training increased plasma adiponectin levels twice as much when compared to moderate intensity [141]. In young lean subjects, on the other hand, acute high-intensity running increased serum adiponectin levels by 10.5%, although reducing its levels in the cerebrospinal fluid by 21.3%. The decrease could be due to an exercise-induced decrease in the blood-brain barrier permeability [142]. Even though, serum and CSF adiponectin levels were positively correlated with the subject’s cognitive performance after exercise [142]. Moreover, we have recently demonstrated that 14 days of AdipoRon treatment restored the cognitive and anxiogenic profile in streptozotocin-induced diabetes to levels equivalent to voluntary wheel running, which was associated with improved hippocampal synaptic plasticity and improved dendritic complexity, spine density, and neurogenesis [77].
The mechanisms mediating the therapeutic effects of adiponectin in AD, diabetes, and obesity are associated with the activation of the AdipoR1/AMPK pathway. Knocking down AdipoR1, but not AdipoR2, blocked the AdipoRon-associated increase in structural plasticity in the in vitro amyloid β-induced model of AD [16]. Moreover, blocking the AMPK activity in vivo before AdipoRon treatment abrogated its effects on spatial memory and neurogenesis in a transgenic animal model of AD [16]. A similar observation was found for Osmotin treatment, which increased hippocampal and cortical expression of AdipoR1 and the activity of its downstream protein targets, including AMPK, Akt, and PI3K in different models of AD [17]. Similarly, When the AdipoR1/AMPK pathway was blocked, Osmotin improvements in terms of neuronal metabolism, plasticity, and cognition were abrogated [17].
As addressed previously, neuronal activation is the main factor driving the high metabolic demands of the central nervous system [48, 49]. It transiently depletes the intracellular energy levels mostly due to the increased activity of ATP-driven ionic pumps intended to reestablish the membrane potential [48, 49]. In agreement with that, sustained neuronal activation can lead to depletion of intracellular energy levels excitotoxic damage [143, 144]. Interestingly, adiponectin counteracted such metabolic stress and was neuroprotective in the kainic acid- [145– 147], and glutamate-induced excitotoxicity [148], ethanol-induced apoptosis [149], and neuronal injury induced by oxygen-glucose deprivation [150]. In the kainic acid-induced excitotoxicity, Osmotin was able to increase neuronal viability and restore hippocampal synaptic plasticity [151]. Furthermore, in an animal model of vascular dementia, AdipoRon treatment increased spatial memory and rescued neuronal survival and axonal integrity in the hippocampal CA1 regions, cortex, and striatum [152].
The AMPK signaling pathway is likely to be involved in such neuroprotective properties [153]. Shortly after glutamate-induced cellular excitation, increased AMPK activity upregulates the membrane translocation of the glucose transporter 3, resulting in increased glucose influx and hyperpolarization of the mitochondria [39]. Such AMPK-dependent restoration of intracellular glucose availability is essential for a rapid recovery of the intracellular ATP levels, which increases neuronal survival and reduces excitotoxic damage [40]. Moreover, we observed that the neurogenic and pro-cognitive effects of AdipoRon treatment in diabetic mice are mediated by increased mitochondrial function [77], suggesting the neuroprotective effects of adiponectin signaling can be mediated by increased mitochondrial activity.
The role of increased central adiponectin signaling in physiological conditions, however, is less clear. In vivo electrophysiological recording of the hippocampal dentate gyrus displayed an increase in LTP formation and prevented long-term depression induction upon 10 minutes of i.c.v adiponectin infusion [154]. Interestingly, adiponectin infusion per se increased the field excitatory postsynaptic potential amplitude [154]. However, local infusion of AdipoRon reduced intrinsic neuronal excitability in the hippocampal dentate gyrus subregion [155] and downregulated spontaneous neuronal activation in the ventral tegmental area [65]. Moreover, 2 h incubation of hippocampal slices with adiponectin impaired LTP in the hippocampal CA1 region [156]. On the other hand, 10 min of AdipoRon application through bath perfusion tended to increase short-term synaptic plasticity in the hippocampal dentate gyrus [77]. Therefore, although adiponectin relevance for synaptic activity has been suggested, it is still not clear what determines the direction of its effect. One possible explanation relies on the dual role of its downstream targets, such as the AMPK signaling pathway, which could also underly the previously mentioned adiponectin paradox
Although AMPK is essential for proper synaptic function and plasticity [39, 40, 55], its overactivity can result in opposite effects. Metformin, a first-line anti-diabetic drug and strong activator of AMPK, blocks the high-frequency stimulation-induced activation of the mTOR pathway and impairs LTP maintenance in the hippocampus [157]. Moreover, 0.1 and 0.3 mM of AICAR incubation leads to increased dendritic length and branching in cultured hippocampal slices, whereas 1 mM leads to a reduction of such parameters [102]. Noteworthy, abnormal AMPK phosphorylation has been found in neuronal substrates of several neurological disorders, including Alzheimer’s and Parkinson’s disease [158]. As previously discussed, AMPK is an inhibitor of the mTOR cascade [38], whose activity is essential for protein synthesis [41] and consolidation of plasticity changes [159]. Nonetheless, if the adiponectin-associated reduction in neuronal excitability and synaptic plasticity under physiological conditions are mediated by AMPK overexpression and if such mechanism is involved in the adiponectin paradox requires further investigation.
CONCLUSION
Neuronal plasticity underlies virtually all functions of the nervous system, being especially relevant for learning and adaptation. Likewise, neuroplasticity impairments are usually reflected in aberrant cognition and mood regulation. The CNS reliance on peripheral energetic resources renders it highly vulnerable to prolonged metabolic oscillations, such as those induced by chronic stress. Novel pro-cognitive and neuropsychiatric drugs are required to consider both aspects to achieve increased efficacy. Adiponectin stands out as a candidate, as experimental research has been showing it can counteract many of the metabolic features associated with impaired neuronal plasticity, resulting in improvements of cognition and psychiatric-like behaviors in several animal models. However, further research is needed. Adiponectin overactivation in physiological conditions can result in reduced neuronal plasticity and the adiponectin paradox observed in clinical settings warns on the necessity for a deeper understanding of adiponectin signaling before therapeutic strategies are advisable.
Declaration: Some sections of the text are part of the first author’s thesis [160] and were adequate to be included in this review.
Footnotes
ACKNOWLEDGMENTS AND FUNDING
The work was supported by funding awarded to SY YAU (Hong Kong Research Grant Council, General Research Fund15100018; 15104019 &15104620); Seed funding support from Mental Health Research Center, and Research Institute for Smart Ageing at Hong Kong Polytechnic University.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.