
Abstract
Precipitous declines in cognitive function can occur in older individuals following a variety of peripheral immune insults, such as surgery, infection, injury, and unhealthy diet. Aging is associated with numerous changes to the immune system that shed some light on why this abrupt cognitive deterioration may occur. Normally, peripheral-to-brain immune signaling is tightly regulated and advantageous; communication between the two systems is bi-directional, via either humoral or neural routes. Following an immune challenge, production, secretion, and translocation of cytokines into the brain is critical to the development of adaptive sickness behaviors. However, aging is normally associated with neuroinflammatory priming, notably microglial sensitization. Microglia are the brain’s innate immune cells and become sensitized with advanced age, such that upon immune stimulation they will mount more exaggerated neuroimmune responses. The resultant elevation of pro-inflammatory cytokine expression, namely IL-1β, has profound effects on synaptic plasticity and, consequentially, cognition. In this review, we (1) investigate the processes which lead to aberrantly elevated inflammatory cytokine expression in the aged brain and (2) examine the impact of the pro-inflammatory cytokine IL-1β on brain plasticity mechanisms, including its effects on BDNF, AMPA and NMDA receptor-mediated long-term potentiation.
INTRODUCTION
Following peripheral immune challenges, older individuals are particularly susceptible to precipitous declines in cognition. A variety of insults, including peripheral surgeries and infections, consumption of unhealthy diets, and injury, have been linked to cognitive impairment in the older population, and deleterious neuroinflammation has emerged as a critical component underlying these deficits. During normal aging, the brain’s resident immune cells –microglia –become sensitized [1], priming them to mount more frequent, prolonged, and exaggerated inflammatory responses [2]. It is hypothesized that when these already sensitized microglia are activated in response to peripheral immune insults the significant neuroinflammatory reaction that results leads to down-stream changes that are damaging to the brain’s synaptic plasticity functions, ensuing in memory deficits and other cognitive impairments. Indeed, expansive research has implicated exaggerated neuroinflammation, in part due to aging-associated microglial priming, in a variety of cognitive deficits following peripheral immune challenge. Here, we examine those studies which investigate the specific role of pro-inflammatory cytokines –namely interleukin 1-beta (IL-1β) –in the development of cognitive decline following peripheral immune activation in the aging population.
BACKGROUND
Immune-to-brain communication & sickness behavior
Normally, the immune and nervous systems are in constant bi-directional communication, and this feedback loop is amplified during an immune challenge. Specifically, pro-inflammatory cytokines (PICs) such as interleukin 1-alpha and beta (IL-1α and β), interleukin 6 (IL-6), and tumor necrosis factor-alpha (TNFα), are released in the periphery following immune stimulation. These cytokines facilitate communication with the nervous system via humoral or neural routes (reviewed extensively in [3–6]), namely: 1) diffusion of cytokines through brain regions which lack a blood-brain barrier (BBB), most notably the circumventricular organs, 2) direct binding of PICs to receptors on the surface of endothelial cells that make-up the BBB, 3) active transport of circulating cytokines across the BBB, or 4) activation of vagus nerve afferents and the sympathetic nervous system by cytokines in the periphery. Within the brain, these immune signals are detected predominantly by microglia that, upon stimulation, secrete additional PICs, thereby potentiating the peripheral immune signals within the central nervous system [7, 8].
Immune-to-brain communication is a critical component underlying “sickness behavior,” an adaptive set of behavioral changes experienced in response to an immune challenge, including fever, lethargy, reduced appetite, and decreased sociability. These behavioral changes reflect the brain’s recognition and interpretation of peripheral immune activation, and actively contribute to the immune response by redirecting energy and enabling the individual to more efficiently resolve infection or injury (reviewed in [9, 10]). It is well-established that cytokines play a key role in mediating the development of this adaptive behavioral response. IL-1β in particular has been implicated in this phenomenon, as administration of IL-1β induces the same behavioral changes that are characteristic of sickness behavior [11]. Further, blocking IL-1β by administering the IL-1 receptor antagonist (IL-1Ra) prevents the physiologic and behavioral effects consistent with the known cytokine-inducer lipopolysaccharide (LPS). Conversely, mice with Type I IL-1 receptor (IL-1R1) knockout remain responsive to the behavioral effects of LPS [12], indicating that IL-1β is not the sole cytokine involved in the mediation of sickness behavior. Administration of TNFα yielded similar results, eliciting the nonspecific symptoms typical of sickness behavior, and central blockade of TNFα prevents such behavioral effects [13–15]. Interestingly though, research indicates that IL-1 signaling is more important than TNF signaling in the context of immune challenge [16]. Additionally, while administration of IL-6 does not elicit the full spectrum of sickness behaviors, it does induce fever and potentiate the behaviorally depressive effect of IL-1β [17]. Further, IL-6 knockout mice are partially protected from the behavioral effects of LPS or IL-1β [18]. These studies implicate PICs in mediating the behavioral processes characteristic of sickness behavior and demonstrate the capacity of certain PICs to modulate behavioral outputs. However, it is important to note that sickness behavior is distinct from cognitive processes such as learning and memory, each having their own unique underlying mechanisms. Of note, sickness behavior is mediated by transient activation of neuroimmune cells, such as microglia and astrocytes, without breakdown of the BBB, infiltration of peripheral immune cells, or other overt pathologies [5, 19].
Microglial sensitization & other age-related neuroimmune changes
Microglia, as myelomonocytic cells, constitute the primary innate immune cells of the brain. These cells exhibit a broad range of functions, most importantly immune surveillance of the brain’s microenvironment for invading pathogens, endogenous danger signals, cellular debris, apoptotic cells, and altered neuronal phenotypes [20]. In the young adult brain, normally surveillant microglia become activated in response to such threats, and undergo physiological and morphological changes, proliferate, and produce PICs [21]. Upon resolution of such threats, alternatively activated microglia secrete anti-inflammatory cytokines and other molecules which facilitate a return to homeostasis [21]. With aging, however, microglia shift towards a more pro-inflammatory phenotype, including upregulation of glial activation markers such as major histocompatibility complex II (MHCII), complement receptor 3 (CD11b), the danger signal HMGB1, and the pattern recognition receptor toll-like receptor 4 (TLR4) [22–24]. Altered morphology and reductions in microglial regulators CD200 and CX3CR1 are also characteristic of these aging cells [2, 23]. Collectively, these changes represent a shift in microglial state from surveillant to sensitized. Under basal conditions, sensitized microglia do not produce increased levels of pro- or anti-inflammatory cytokines. However, upon stimulation, sensitized microglia are primed to mount more robust and prolonged immune responses, including exaggerated production of PICs [25–27]. Importantly, a broad array of stimuli –bacterial and viral infections, surgery, psychological stress, brain injury –have been shown to activate these cells and induce exaggerated and prolonged neuroinflammatory responses in older rodents as compared to younger ones.
It is unclear why the brain’s innate immune cells experience this age-related sensitization. However, multiple aspects of the immune system undergo aging-associated changes. Briefly, in aging, the innate immune system is generally associated with dysregulated or heightened responses in both the periphery and the nervous system, leading to chronic low-grade inflammation [28, 29]. In contrast, the adaptive immune system is generally suppressed by aging, as evidenced by reduced production of B and T cells and diminished function of mature lymphocytes, including reduced abilities to fight infections and elicit vaccine responses [29–32]. While it remains to be determined exactly why the immune system experiences these changes, one thought is that chronic inflammation induces compensatory immunosuppressive mechanisms in aging [33, 34]. Yet, others suggest that increased innate immune activity is actually compensating for reduced adaptive immune activity [30, 35, 36]. The latter could hold true for microglia as well – as myelomonocytic cells, the brain’s innate immune cells may undergo adaptive sensitization to account for reduced adaptive immune responses, which could make it more likely for harmful pathogens to invade the central nervous system, particularly when paired with increased BBB permeability in older age [36, 37]. Indeed, normal aging is associated with alterations to the bi-directional routes of immune-brain communication [38, 39].
As previously described, both direct binding of PICs to receptors on the surface of endothelial cells that make-up the BBB and active transport of circulating cytokines across the BBB constitute a critical means of immune-brain communication [3–6]. Thus, physiological states which involve deterioration of the BBB might allow for increased levels of inflammatory mediators to enter the central nervous system and may therefore induce an exaggerated neuroimmune response. Indeed, both aging and aging-related cognitive disorders have been associated with increased BBB permeability and leakage [38–40]. Intriguingly, several studies have indicated that the hippocampus –a region critical to memory function –is particularly vulnerable to this age-related BBB deterioration [41, 42]. Additionally, the mechanisms of transport across the BBB also appear to experience age-related alterations. For example, a recent study demonstrated a shift from ligand-specific receptor-mediated transport to non-specific transcytosis, enabling transport of proteins which lack a canonical transport receptor –and thus are unlikely to enter the CNS in young brains –across the BBB [43]. The same study reported a reduction in pericyte coverage along the neurovascular unit, consistent with other reports of changes to BBB structure and composition [43, 44]. Dysfunctional endothelial transport has also been associated with aging-related BBB breakdown [39]. Additionally, astrocytes, which in addition to their own neuroimmune functions play a critical role in maintaining the BBB, also experience aging-related functional changes and may contribute to breakdown of the BBB [45]. While these are all important considerations with regard to age- and neuroinflammation-associated cognitive impairments, and which likely contribute to aging-associated neuroinflammatory priming, they will not be discussed further here in an effort to keep this review narrow in scope.
Inflammation & cognitive decline
Clinically, peripheral insults such as surgery and infection are known to elicit detrimental cognitive deficits in older individuals (Fig. 1). Preclinically, we and others have demonstrated that an array of surgeries, such as various abdominal, orthopedic, and cardiac surgeries, result in exaggerated neuroinflammation and, consequentially, cognitive impairment, in aged but not young adult rodents [46–51]. Similarly, peripheral infections, including E. coli, influenza A, and human immunodeficiency virus (HIV), have been shown been associated with memory impairments disproportionately in aged systems as compared to young adult ones. Excessive neuroinflammation resulting from normal aging-associated microglial priming is implicated in the underlying processes [52–54]. Additionally, consumption of diets high in fat and sugar have been shown to impact cognition. In fact, high fat diet (HFD) consumption is sufficient to cause cognitive deficits in aged rodents, but not young adults, in as a little as 3 days [55]. One mechanism of diet’s effects on cognition is its ability to induce a robust inflammatory response in the aged brain, in part by activating TLR4 on microglia (Fig. 1) [55–62]. Indeed, elevated expression of PICs and impaired phagocytic activity by microglia are seen to parallel HFD-induced memory deficits in aged rodents [55, 57]. Furthermore, neuroinflammation has been implicated in a variety of aging-associated cognitive diseases, most notably Alzheimer’s disease and other dementias, as well as in the pathology of traumatic brain injury [63–68].
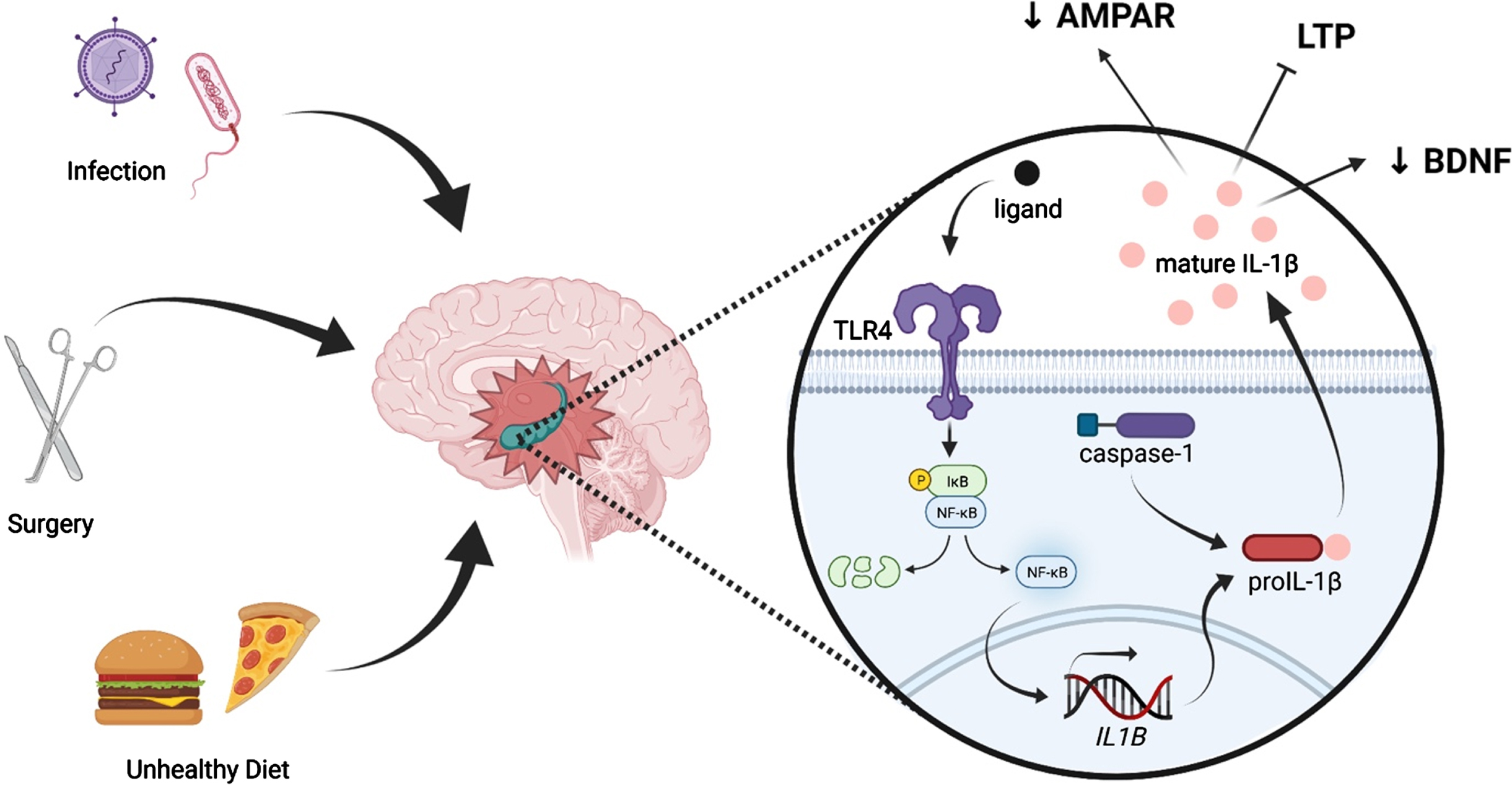
Peripheral immune challenges such as infection, surgery, and consumption of an unhealthy diet can evoke neuroinflammation, especially in the hippocampus. Such insults are known to activate the pattern recognition receptor TLR4, leading to activation of NF-κB and, ultimately, production of immature IL-1β (pro-IL-1β). Pro-IL-1β is cleaved by caspase-1, an inflammasome component, into mature IL-1β, which is released into the extracellular fluid. IL-1β elicits multiple effects on synaptic plasticity-related processes, including suppression of BDNF production, reduction of AMPAR membrane expression, and inhibition of LTP.
Fascinatingly, multiple inflammatory challenges, or “hits,” are capable of potently exacerbating such cognitive deficits. In what has become known as the “multi-hit theory” of neuroinflammation, a “hit” can be defined as an immune challenge which elicits a neuroinflammatory response. This is in contrast to low-grade inflammatory events, which induce a local cytokine response without triggering their activation in the brain [69]. In the above examples, aging is a single inflammatory “hit,” and insults such as surgery, infection, or injury act as second “hits,” further elevating the expression of neuroinflammatory molecules. As the number of immune hits increases, so too does the neuroinflammatory sequalae and the detriments to cognition. For example, we have previously shown that in aged (but not young adult) rats, surgery induced hippocampal-dependent memory impairments lasting 4 days [25]. Of note, the addition of morphine –which confers an inflammatory affect in addition to its analgesic properties (i.e. a third inflammatory “hit”) –to the post-surgical regimen robustly extended the memory deficit from just 4 days without morphine to at least 8 weeks with morphine [46]. This long-lasting cognitive dysfunction was associated with exaggerated neuroinflammation only in the aged animals who received both surgery and morphine treatment [46]. Importantly, multiple inflammatory insults do not just increase susceptibility to cognitive dysfunction in aging. Indeed, it has been demonstrated that multiple inflammatory “hits” in young rodents can induce changes that make them appear immunologically and behaviorally similar to aged rodents.
Püntener and colleagues, for example, showed that in 8 week old mice, systemic infection with the bacterium Salmonella typhimurium induced prolonged cytokine synthesis in the brain and primed microglia to subsequent inflammatory challenges [70]. The group observed significant upregulation of microglial CD11b and CD68 7 days post-infection [70]. At 21 days after infection, significant elevation of the PICs IL-1β, IL-12, and IL-6 was also observed [70]. Importantly, Püntener et al. demonstrated that this ‘primed’ microglial phenotype made the 8 week old mice vulnerable to subsequent inflammatory challenges, as intrahippocampal administration of LPS 4 weeks after infection led to a heightened inflammatory response when compared to control groups [70]. Similarly, Frank et al demonstrated that stress (inescapable shock) exacerbated the inflammatory response of hippocampal microglia from young adult rats to LPS ex vivo 24 hours after exposure to the stressor [71]. A similar phenomenon was observed by Wohleb and colleagues in a repeated social defeat stress paradigm. The group showed that repeated social defeat induced an inflammatory profile in microglia from young adult mice and that this pro-inflammatory status resulted in exaggerated production of inflammatory cytokines following ex vivo stimulation with LPS [72]. Notably, Sobesky et al., showed that, in young adult rats, consumption of a high-fat diet for 3 days elevated expression of microglial priming markers associated with aging [73]. Further, immune challenge with a low dose of LPS following short-term HFD consumption potentiated the neuroinflammatory response and elicited an impairment in long-term memory formation [73], similar to what has been observed in aged rats [55].
Given the prevalence of neuroinflammation in memory impairments and other cognitive deficits which disproportionately affect aged individuals, it is critical that we understand the mechanisms by which peripheral immune activation incites these effects. As described, PICs are a critical component of the immune response elicited by peripheral immune challenges. These peripheral inflammatory molecules, most prominently IL-1β, initiate a signaling cascade within the central nervous system, including central production of PICs. Within the brain parenchyma, these cytokines directly and indirectly activate microglia, thereby propagating the neuroimmune response. This cascade is critical to the development of adaptive sickness behaviors such as lethargy, reduced appetite, and decreased sociability, which help facilitate energy redirection and enables the inflicted individual to more efficiently resolve the immune challenge. However, aging is associated with microglial sensitization, which primes those cells to mount more frequent and exaggerated neuroimmune responses. This aging-associated amplification of neuroimmune responses has been associated with a plethora of cognitive deficits following peripheral immune challenge, including infections, surgery, and injury. How, though, do these normally beneficial inflammatory molecules, which are critical to fighting infection and repairing injury, elicit such detrimental effects as cognitive deterioration? As described above, PICs –namely IL-1β and TNFα –are required for the development of sickness behavior. This strongly implicates a role for PICs in modulating cognitive processes such as social behavior and memory and provides a framework by which excessive production of PICs may turn deleterious and elicit maladaptive effects on cognition & the underlying processes of synaptic plasticity. Below, we highlight recent studies that examine the specific mechanisms by which PICs –in particular, IL-1β –impair brain plasticity to elicit detrimental effects on an array of cognitive processes.
IL-1 SIGNALING, SYNAPTIC PLASTICITY, & MEMORY
IL-1 signaling is required for LTP & hippocampal-dependent memory
As aforementioned, IL-1β is a critical mediator of neuroimmune responses and related sickness behaviors. Relevantly, central IL-1β and its receptor IL-1R, as well as the receptor antagonist IL-1Ra, are regionally selective and especially prominent within the hippocampus [74–77], a brain region critical to contextual and spatial memory. The localization of IL-1 to this region suggests it and its regulatory molecules may have a role in modulating hippocampal-dependent memory and provides a mechanism by which aberrantly elevated IL-1β might elicit detrimental effects to such memory functions. Long-term potentiation (LTP) is a form of synaptic plasticity and involves synaptic activity patterns which elicit persistent increases in signal transmission between a set of neurons. This form of synaptic plasticity has been shown to underlie memory formation, including in the hippocampus. It has been known for some time that IL-1β is physiologically increased during LTP, and that blockade of the IL-1R with IL-Ra results in impaired LTP [78], suggesting that IL-1β is necessary for functional LTP. Indeed, mice who lack IL-1R exhibit no LTP within the dentate gyrus and CA1 regions of the hippocampus. This is paralleled by impaired hippocampal-dependent memory (Morris water maze, contextual fear conditioning) [79]. This may be because IL-1β appears to be required for memory consolidation [80].
Indeed, a number of studies in recent years have demonstrated the necessity of IL-1β for hippocampal-dependent memory. Labrousse and colleagues found that mice deficient for P2X(7) receptor (P2X(7)R), which is required for endogenous synthesis of IL-1β, were impaired in a hippocampal-dependent spatial memory task [81]. Interestingly, P2X(7)R-/- mice were unimpaired in an object recognition task, which is not dependent on the hippocampus [82]. Similarly, IL-1β (as well as IL-6 and TNFα) in hippocampal CA1 has been implicated in proper contextual fear memory, another hippocampal-dependent memory task [82]. Interestingly, recent evidence suggests that this necessity of IL-1β for proper memory function may be dependent on age. A 2017 study from Takemiya and colleagues found that spatial memory was significantly impaired in young mice (3mo) with either IL-1β or IL-1R KO, whereas adult (6mo) IL-1β or IL-1R KO mice were unimpaired [83]. These results suggest the critical role of IL-1β in memory function may be limited to young rodents. Indeed, the vast majority of studies which investigate normal learning and memory are performed in younger rodents.
Aberrantly elevated IL-1β is associated with memory impairment
Conversely, a number of studies indicate that significantly elevated levels of IL-1β inhibit LTP and contribute to hippocampal-dependent memory dysfunction [84, 85]. For example, in hippocampal slices, IL-1β has been shown to inhibit LTP [86, 87]. In hippocampal neuron cultures, IL-1β was similarly found to inhibit LTP [88]. This dysfunction was associated with inhibition of AKT/mTOR signaling (which is required for LTP induction and maintenance [89]) and dysregulation of AMPA, a receptor protein critical to synaptic transmission and plasticity. In vivo, Barrientos et al demonstrated that the combination of aging and an immune challenge (peripheral E. coli infection) elicited a robust neuroimmune response, including exaggerated production of IL-1β, which was mirrored by deficits in the consolidation of hippocampal-dependent memory [90], underlying this memory dysfunction was specific disruption of theta-burst late-phase LTP in the CA1 region of the hippocampus [91]. These deficits were abrogated by central administration of IL-1Ra [91]. Similarly, post-operative cognitive decline is characterized by significant hippocampal-dependent memory impairment and exaggerated neuroinflammation, including elevated production of IL-1β, in aged rodents, and treatment with IL-1Ra ameliorates such deficits [25, 46].
A 2017 study by Tomasoni and colleagues examined the effects of hyperactivation of the IL-1 signaling pathway by knocking out the IL-1R 8 (IL-1R8), which interferes with the activation of the IL-1R signaling pathway [92]. The group found that KO of IL-1R8 resulted in gross altercations to synaptic architecture of hippocampal neurons, including decreased expression of the post-synaptic protein post-synaptic density 95 (PSD95), which correlated with reductions in PSD95 puncta density and size. Consequentially, reductions in miniature excitatory post-synaptic currents were also observed. Functionally, primary hippocampal cultures from IL-1R8 KO mice were unable to undergo synaptic plasticity following exposure to an established chemical LTP protocol. Interestingly, administration of IL-1Ra to IL-1R8 KO neurons restored LTP functionality. In a partial delineation of the underlying mechanisms, Tomasoni et al demonstrated that the PI3K/AKT/mTOR signaling pathway underlies the dysfunctional effects of hyperactivated IL-1 signaling. Indeed, the PI3K/AKT/mTOR pathway is required for LTP induction and maintenance [89]. The group found that treatment of IL-1R8 KO neurons with the known mTOR inhibitor rapamycin increased PSD95 expression and reversed the dendritic spine abnormalities observed in untreated cells [92]. Inhibition of PI3K/AKT signal transduction similarly ameliorated the structural dysfunction [92]. These studies suggest that hyperactivation of IL-1 signaling leads to upregulation of the mTOR pathway, resulting in defects to dendritic spine morphology and related mechanisms of synaptic plasticity, namely LTP.
Interestingly, aging is associated with a heightened susceptibility to cognitive decline related to elevated IL-1β [25, 46]. Prieto and colleagues investigated this apparent vulnerability of the aged hippocampus to IL-1β. By tracking LTP within synaptosomes, the group demonstrated that IL-1β impairs LTP directly at the synaptic level, and that aged hippocampal synapses are especially sensitive [93]. Further, it was found that this age-dependent sensitivity to IL-1β is due to reconfiguration of IL-1R subunits, specifically characterized by a shift in the ratio of AcP (pro-inflammatory) and AcPb (pro-survival) subunits [93]. The authors speculate that aging-related increases in IL-1β induce this shift in IL-1R arrangement. Indeed, IL-1R is differentially expressed by age, with aged rodents expressing elevated basal levels of the cytokines, in some strains [94–97]. Peripheral immune challenges have been shown to “prime” the learning-associated increase in IL-1β, resulting in deleterious expression. For example, it has been shown that consumption of a pro-inflammatory HFD is associated with potentiated hippocampal IL-1β production 2 hours after a learning experience (in this case, involving an aversive foot shock) and, consequentially, impaired hippocampal-dependent contextual memory. Central administration of IL-1Ra prevented this dysfunction [98].
IL-1β suppresses BDNF production
Brain-derived neurotrophic factor (BDNF), as a neurotrophin, plays a critical role in the differentiation, development, and survival of certain neuronal populations. BDNF is also particularly important to synaptic plasticity processes which underlie hippocampal-dependent memory consolidation, as BDNF can directly facilitate LTP (reviewed extensively in [99, 100]). Thus, factors which modulate BDNF can negatively influence functions downstream of functional LTP. In recent years it has been evidenced that neuroinflammation plays a role in regulation of BDNF expression. Tong and colleagues demonstrated that IL-1β specifically suppresses BDNF signal transduction in neuronal cultures and that IL-1β regulates BDNF signaling in hippocampal slices [101, 102]. The group specifically posited that IL-1β impairs phosphorylation of insulin receptor substrate 1, a protein which couples BDNF to its effector TrkB, thereby preventing activation of the PI3K pathway (which is implicated in LTP) [101]. A 2017 study expanded on this work and revealed that, in primary hippocampal neurons, IL-1β treatment reduced the retrograde transport of signaling endosomes. Functionally, this deficit correlated with inhibition of Erk5, which is normally activated by BDNF following retrograde endosome trafficking [103]. Importantly, Erk5 is known to promote neurogenesis, particularly in the hippocampal dentate gyrus, and plays a critical role in long-term memory maintenance [104–106]. It should also be noted that endocytosis is a critical component of a variety of signaling cascades, including other neurotrophins, and it is thus possible that IL-1β’s effect on endosomal trafficking is not specific to BDNF, although this remains to be examined.
IL-1β modulates AMPA & NMDA receptors
Glutamatergic neurotransmission is understood to play a critical role in various forms of synaptic plasticity, including LTP. In particular, glutamate signaling via the ionotropic glutamate receptors 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl) propanoic acid (AMPA) and N-methyl-D-aspartate (NMDA) is especially important for learning and memory function [107, 108]. As such, both AMPA and NMDA receptors have been studied extensively in the context of memory impairment, although their role in neuroinflammatory-mediated cognitive deficits is of more recent interest. Coogan and O’Connor first demonstrated that IL-1β inhibits NMDA, but not AMPA excitatory post-synaptic potentials and LTP in the rat dentate gyrus region of the hippocampus [109]. Since then, a number of studies have implicated impaired LTP and dysregulated glutamatergic signaling in the detrimental effects of neuroinflammation and, specifically, IL-1β. Min and colleagues have shown that chronic neuroinflammation induced by LPS treatment for 4 weeks (via intraventricular infusion) reduced both NMDAR-dependent and NMDAR-independent LTP in the CA1 [110]. An intriguing study from Li and colleagues found that caspase-1/IL-1β signaling underlies alterations to AMPA receptors and behavioral deficits observed in a model of chronic stress. Specifically, the group demonstrated that inhibition of caspase-1/IL-1β signaling prevented the anxiety- and depressive-like behavioral abnormalities and averted the characteristic decrease in AMPA receptor membrane expression within the hippocampus [111].
Recent work from Taoro-Gonzalez and colleagues investigated the mechanistic underpinnings of cognitive dysfunction in a hyperammonemic rat model of hepatic encephalopathy. Since neuroinflammation is known to modulate the development of this hippocampal memory impairment, the group investigated the effects of IL-1β on AMPA receptor subunits in the hippocampus [112]. Their studies demonstrated that hyperammonemia resulted in elevated expression of hippocampal IL-1β as well as reduced GluA1 and increased GluA2 membrane expression [112]. Blockade of IL-1 signaling with IL-1Ra prevented these changes to membrane expression of the AMPA receptor subunits [112]. Activation of the IL-1 receptor is known to increase phosphorylation and activity of Src kinase, which is capable of mediating GluA1 and GluA2 expression [112]. As such, the group investigated the role of Src signaling in the observed changes. Indeed, inhibition of Src was also found to prevent the changes in membrane expression of GluA1 and GluA2 in hyperammonemic rats [112]. Further mechanistic studies from Taoro-Gonzalez revealed that IL-1 signaling enhanced Src, which increased the phosphorylation and membrane expression of GluN2B, an NMDA receptor subunit [112]. This in turn led to the activation of MAP kinase p38 which reduced PKCζ activity and, ultimately, resulted in reduced phosphorylation and increased membrane expression of GluA2 [112]. Via a different pathway, increased Src activity was found to activate PKCδ, enhancing phosphorylation of GluN2B and thus reducing membrane expression and phosphorylation of CaMKII and GluA1 [112]. Also in 2018, a study led by Tong similarly revealed a Src-mediated mechanism by which IL-1β reduced the membrane expression of GluA1 following chemical LTP [113]. Together, these studies suggest a regulatory role for IL-1β in surface expression of various AMPA and NMDA receptor subunits, which may underlie cognitive deficits associated with neuroinflammation.
Taoro-Gonzalez, Tong, and colleagues identified several Src-mediated mechanisms by which neuroinflammation (specifically IL-1β) can modulate glutamatergic signaling within the hippocampus by altering the membrane expression of AMPA and NMDA receptor subunits. Interestingly though, TNFα confers the opposite effect, whereas IL-1β reduces the membrane expression of GluA1 and increases that of GluA2, TNFα has been shown to increase GluA1 and reduce GluA2 membrane expression [114–116]. These conflicting findings indicate that different types of neuroinflammation (i.e. different sources, persistence, dynamics, or strengths) may elicit different effects on AMPA receptor subunits, although it should be noted that in both cases glutamatergic neurotransmission is altered, impairing hippocampal LTP. However, given the age-related changes to IL-1β expression and IL-1 receptor sensitivity discussed previously, it may be that aged animals are more likely to experience IL-1β-mediated alterations to the AMPA receptor subunits, and this variation could partly underlie the susceptibility of older individuals to cognitive impairments.
IL-1β and oxidative stress, neurogenesis, and BBB permeability
Multiple mechanisms have been identified as contributing to the detrimental effects of inflammation. Here we have focused on the effects of PICs, namely IL-1β, on synaptic plasticity-related processes. However, it should not be overlooked that PICs elicit an array of other effects which have been associated with cognitive dysfunction. For example, PICs such as IL-1 have a profound effect on oxidative stress and, in models of sepsis, inflammation-induced pain sensitivity, chronic cerebral hypoperfusion, infection and others, blockade of IL-1 or other PICs has been shown to ameliorate both the associated increases in oxidative stress as well as the related neurological and cognitive deficits [117–120]. However, oxidative stress alone has been shown to induce neuroinflammation and cognitive dysfunction [121], and so it is still unclear which is the true instigating culprit, although it remains a possibility that this depends highly on the nature of the immune insult or condition. Similarly, elevated PICs, including IL-1, have been associated with reduced neurogenesis, particularly in the hippocampus [122–127]. Furthermore, this effect can be ameliorated by blockade of IL-1 [115, 122, 124]. We previously described the effects of aging on BBB breakdown, but it is important also to note that elevated PICs can elicit similar effects, increasing BBB permeability [128–130]. This is particularly relevant to consider as it may be that production of inflammatory cytokines following immune challenge actually potentiates age-related BBB breakdown, contributing to the exaggerated neuroinflammation observed in older individuals.
CONCLUSIONS
Presently, we described only a sampling of research implicating IL-1β and other PICs in the pathology of acute cognitive decline. In particular, we focused on research that contributed to delineating the apparent vulnerability of aged individuals to such cognitive deterioration, especially following peripheral immune challenges. Indeed, peripheral immune insults such as surgery or infection initiate a robust inflammatory response that is translated to the central nervous system, stimulating local production of pro-inflammatory molecules within the brain parenchyma. Normally this production of PICs is beneficial, as it initiates classic, adaptive “sickness behavior,” including fever, reduced sociability, and fatigue. However, aging is associated with neuroinflammatory priming (particularly microglial sensitization), whereby the brain’s immune cells –namely microglia –elicit exaggerated inflammatory responses upon stimulation, including heightened production of PICs. This overt and deleterious neuroinflammatory response has been shown to underlie a variety of aging-related cognitive deficits, including those experienced following surgery, infection, and even consumption of an unhealthy diet.
As described in the present review, IL-1β, a critical PIC, has been shown to be necessary for proper synaptic plasticity and memory function. Conversely, aberrantly elevated IL-1β and hyperactivation of the IL-1R signaling pathway have been shown to elicit impairments to LTP and long-term memory. These converging lines of research suggest that optimal IL-1β levels may be necessary for proper synaptic plasticity and hippocampal-dependent memory, and that deviations above or below that ideal may be detrimental to cognition (Fig. 2). So why, then, are aged individuals particularly vulnerable to IL-1β-related cognitive deficits? A number of studies indicate that IL-1β is naturally increased in the aged brain, and recent work suggests that this increase may lead to reconfiguration of IL-1R subunits in a manner that promotes inflammation. Aging itself is also associated with chronic low-grade inflammation. It may be that the combination of aging –with its elevated basal inflammation and sensitized immune cells –and a peripheral immune challenge synergize to produce robustly exaggerated levels of PICs, including IL-1β. At such heightened levels, the deleterious effects of aberrantly elevated IL-1β –reduced BDNF production, alterations to NMDA and AMPA receptors, and consequential deficits to LTP –may be too much to overcome, creating the perfect “cytokine storm,” at which point overt negative consequences to cognitive processes like long-term memory occur.
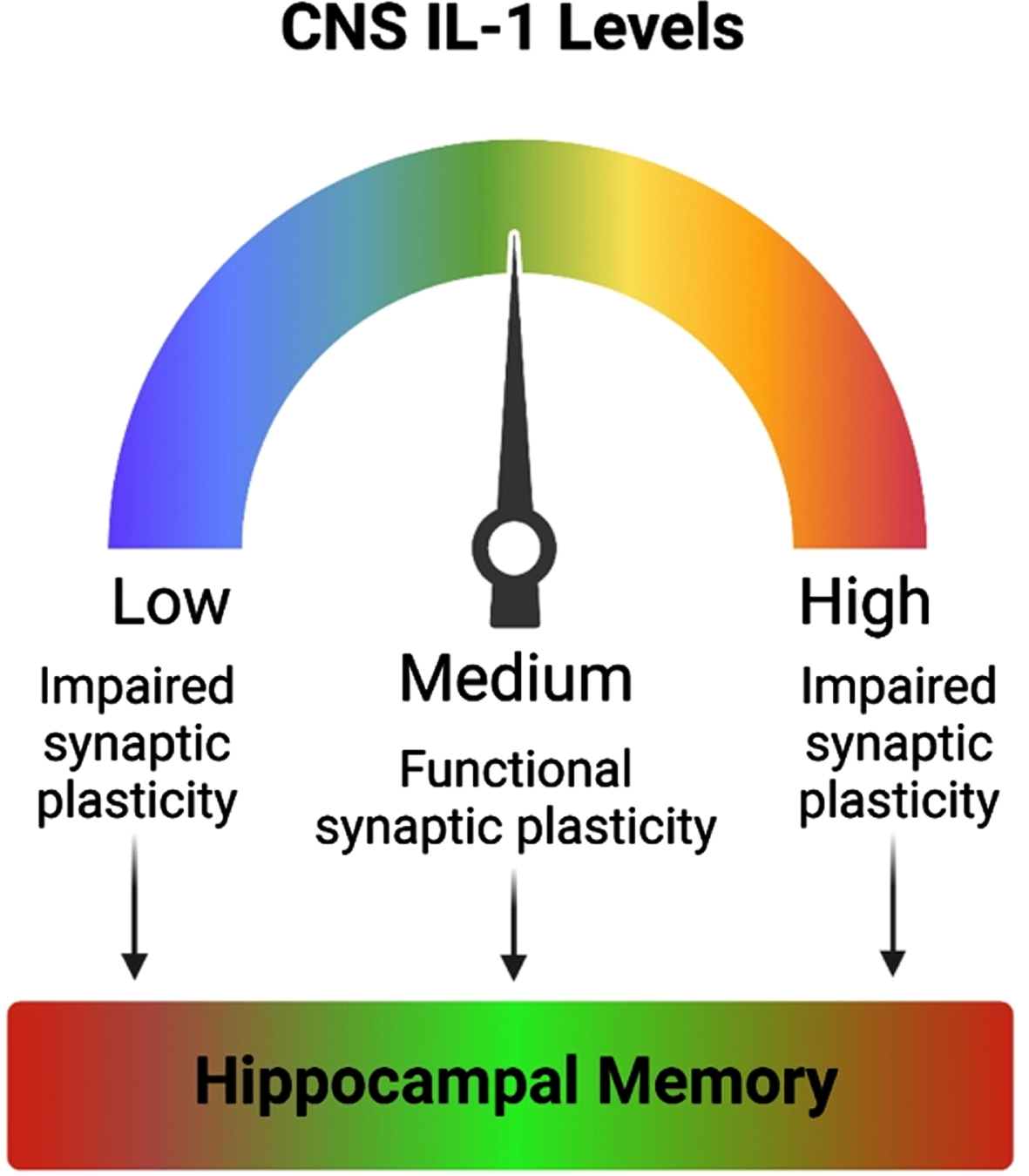
Optimal levels of IL-1 are needed for proper memory function. Inhibition of IL-1 signaling results in impaired synaptic plasticity. Likewise, exaggerated levels of IL-1 or hyperactivation of the IL-1 signaling pathway also impairs synaptic plasticity. Both aberrantly reduced and elevated IL-1 result in memory impairments. Thus, optimal levels of IL-1 are required for proper synaptic plasticity and hippocampal-dependent memory function.
In less than 10 years approximately a quarter of the U.S. population will be over 65 [131]. With advanced age being the strongest risk factor for disruptions in memory function, future research needs to emphasize better understanding the central effects of peripheral immune activation in both males and females [132]. In particular, delineating the effects of immune challenge on plasticity-related processes. Seeking both pharmaceutical and non-pharmaceutical (behavioral) treatment strategies to reduce the deleterious effects of exaggerated neuroinflammation without preventing the beneficial aspects of central immune function is crucial given that people are continuously living longer. Many strides have been made in this regard (reviewed in [133]). For example, it is now well-established that diet can extensively modulate immune function. As discussed here, consumption of diets high in fats and sugars can induce a robust pro-inflammatory phenotype [55–61]. However, certain dietary regimens can elicit an anti-inflammatory response. In particular, consumption of foods with high amounts of antioxidants, such as nuts and berries, as is consistent with a Mediterranean-style diet, has been associated with anti-inflammatory benefits and improvements in cognitive function [134, 135]. Likewise, exercise has profound anti-inflammatory effects in addition to its effects on synaptic plasticity, such as increasing BDNF, resulting in protection against neuroinflammation-related memory impairments [55,136]. Importantly, these positive effects of both diet and exercise on inflammation and cognition exist even when they are initiated in older age [133]. Thus, lifestyle modifications which elicit anti-neuroinflammatory effects pose significant potential to blunt aging-related increases in neuroinflammation and thus protect against inflammation-associated cognitive dysfunction.
Footnotes
ACKNOWLEDGMENTS
This work is supported in part by grants from the National Institute on Aging RF1AG028271 and R03AG067061 to R.M.B.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.