
Abstract
Tyro3, Axl and Mertk are members of the
Keywords
THE TAM RECEPTORS, THEIR LIGANDS AND TAM SIGNALING
Tyro3, Axl and Mertk collectively termed TAMs are tyrosine kinase cell surface transmembrane receptors capable of sensing extracellular ligands, phosphorylating and activating diverse downstream signaling pathways which are necessary for maintaining different aspects of cellular physiology [1]. All TAMs are activated by growth-arrest-specific-protein 6 (Gas6), which binds Axl with highest affinity [2–5]. A structurally homologous ligand Protein S (PROS1) can activate Tyro3 and Mertk but not Axl [6, 7]. The structure of TAM ligands and receptors has been extensively described in recent reviews [1, 8, 9]. Both TAM receptors (TAMRs) and their ligands are expressed across a myriad of cells, influencing various systems including in the immune, reproductive, vascular, and the central and peripheral nervous system (CNS and PNS, respectively). Co-expression of two TAMRs is common, allowing functional redundancy, but this co-expression also provides diverse and regulated functions. TAMRs play pivotal roles in cellular decisions such as cell proliferation and survival, and are instrumental in cell function, with efferocytosis (the phagocytic uptake of apoptotic cells) and immune regulation as key features. Altogether, both arms are necessary for homeostatic regulation and tissue repair [10, 11].
Loss of function mutants in mice via the deletion of TAMRs and ligands have elucidated the diverse roles undertaken by this pathway. Single, double and triple knockout mice are born without obvious developmental defects [12]. Triple knockout mice deficient for all three TAM receptors are viable and healthy for the first few post-natal weeks, but consequently develop postnatal retinal degeneration, autoimmunity and male sterility [12–18], with increased apoptosis and cellular degeneration in the hippocampus and the cerebellum [12]. Axl deletion alone induced increased vascular permeability and Mertk-/- mice are blind due to photoreceptor degeneration [16]. Therefore, TAM function is necessary for maintaining healthy adult tissues. Gas6 knockouts are viable, do not develop retinal degeneration, autoimmunity or infertility and are without any reported neural phenotypes. Gas6 deficiency was reported to protect against thrombus formation and liver inflammation [19, 20]. PROS1 is recognized for its essential function as a blood anti-coagulant, and its systemic deletion is embryonically lethal due to coagulopathy, extensive haemorrhage and necrosis throughout the embryo [21]. The role of PROS1 as a TAM agonist has recently emerged from studies of numerous cell specific conditional PROS1 deletions in various tissues [22–26]. Within the nervous system, deletion of Pros1 was reported in neural lineage, including neural stem cells using Nestin-Cre [24, 25], and in myeloid cells [22], also targeting microglia [25].
Despite the functional redundancy of TAMRs and ligands seen in the immune system, the downstream diversity in cellular responses is quite remarkable and is attributed to activation of distinct intracellular signaling pathways activated in a context-specific manner. Gas6/Axl signaling can affect cell survival, growth and proliferation via the activation of PI3K-Akt, ERK1/2 and MAPK pathways [27]. PROS1-mediated Tyro3 signaling activates the PI3K-Akt pathway which protects cortical neurons from NMDA-excitotoxicity induced apoptosis [28]. In the retina, Gas6 and PROS1 activation of Mertk stimulates phagocytosis of the outer segments of photoreceptors, which is essential in supporting healthy retinal tissue [7, 26]. The dysregulation of TAM signaling is observed in a plethora of pathological processes including cancer progression, chronic inflammation and autoimmune disease [8, 17, 29–32]. In this review we will discuss the biological function of TAM receptors and their ligands in the nervous system and explore the dysregulation of TAM receptors in neuroinflammatory and neurodegenerative conditions and diseases.
TAMRs AND THEIR LIGANDS IN THE NERVOUS SYSTEM
All members of the TAM family are expressed in the developing and adult nervous system. Of the receptors, Tyro3 is the most widely expressed in the nervous system, expressed embryonically but its expression surges postnatally and into adulthood, concurrently with synaptogenesis [33]. Analysing the developing rat brain revealed high expression of Tyro3 in the cerebral cortex, hippocampus, cerebellum, amygdala, piriform cortex, olfactory bulbs, subventricular zones, forebrain, midbrain and hind brain [33, 34]. Furthermore, neurons, glia, astrocytes and oligodendroglia all express Tyro3 [33]. Axl and Mertk are expressed in the nervous system rather constantly during development, however, at lower levels [33]. Axl is expressed in the cerebellum and hippocampus by oligodendrocytes, astrocytes and microglia and by Schwann cells in the PNS [33, 35, 36]. Mertk is detected in astrocytes and oligodendrocytes and is highly expressed by microglia [33, 35–39].
Among the ligands Gas6 is secreted by neurons and endothelial cells, produced in the cerebral cortex, hippocampus, cerebellum, midbrain and thalamus and expressed by microglia, astrocytes and neural stem cells [5, 34, 40]. PROS1 is expressed in neural stem cells, Schwann cells, neurons, astrocytes and microglia [5, 24, 25, 38]. Thus, many cells within the nervous system express one or more TAMRs and secrete one or both of the cognate ligands PROS1 and Gas6, allowing either autocrine and/or paracrine signaling, which lead to cell autonomous as well as non-cell autonomous phenotypes. These co-expression patterns allow tight modulation of various physiological and cellular process contributing to health. As mentioned, TAMRs are expressed in many cells of the developing nervous system, but neural development is not grossly affected in mice lacking expression of all three receptors [12, 24]. Moreover, their specific roles in different cell types is not yet understood, mainly due to the lack of mouse models allowing cell-specific deletion of the different TAM components. The first conditional deletion of Mertk in microglia was recently published [36].
TAMS IN NERVOUS SYSTEM DEVELOPMENT AND HOMEOSTASIS
Typical to any physiological system, and the CNS is no exception, many cellular processes and regulatory functions are critical for proper development and to maintain homeostasis. Dysregulation of these processes may disrupt neural development, homeostasis and cause life-long complications. Within the CNS, TAM signaling is involved in regulating development and homeostasis via both autocrine/cell-autonomous and paracrine/cell non-autonomous modes of action, described below.
TAM RECEPTORS IN THE DEVELOPMENT OF GnRH NEURONS
Gonadotropin-releasing hormone (GnRH) neurons regulate reproduction processes by secretion of GnRH from the pituitary into the hypophyseal portal bloodstream. GnRH activity is very low in childhood, increasing as puberty and adolescence strikes. The GnRH neurons originate in the embryonic nasal placode and migrate along the olfactory axon fibres to the pre-optic area and hypothalamus during development [41]. The migration of GnRH neurons must follow a precise pathway to reach their target projections to induce gonadotropin secretion. Both Tyro3 and Axl are necessary for GnRH neuronal migration and survival, and their loss leads to reproductive malfunctions [42, 43]. Axl is involved in GnRH neuronal cell migration via the activation of the PI3K-Ras pathway and P38 MAPK [44, 45]. Gas6/Axl activation of ERK and PI3K/Akt pathways is key to the survival of migrating GnRH neurons by protecting them from programmed apoptosis [45] and directing GnRH migration to the ventral forebrain [43]. Deletion of Axl and Tyro3 in mice led to a significant loss of GnRH neurons, subsequently impairing the sex hormone induced gonadotropin surge which lead to abnormalities in the estrous cycle of female mice [43]. Furthermore, the loss of Gas6 at any stage embryonically and in adulthood results in a reduced GnRH neuronal population, delays vaginal opening and sexual maturation [46]. GnRH development occurs embryonically, but numerous processes which involve TAM signaling that are developmental in nature- also occur postnatally. Among these are adult neurogenesis, synaptogenesis, synaptic pruning and myelination, discussed below.
TAM SIGNALING IN NEUROGENESIS
Neurogenesis is a process by which neurons are generated from neural stem cells and progenitor cells. Massive neurogenesis occurs prenatally, and the process continues to a lesser extent throughout adulthood [47, 48]. Adult neurogenesis is restricted to the subgranular zone (SGZ) of the dentate gyrus, and the subventricular zones (SVZ), lining the lateral walls of the lateral ventricles. The newborn neurons in adult neurogenesis are generated from a pool of stem cells found in these two regions [49, 50]. Over their lifetime neural stem cells are faced with many binary decisions: to stay quiescent or to proliferate, to differentiate or to self-renew, and finally once differentiated whether to adopt glial or neural fates [51]. These decisions are regulated by cell intrinsic as well as cell to cell interactions and complex external molecular cues [49, 51].
All TAMRs are expressed by neural stem cells (NSCs) [52], and play a vital role in NSC proliferation, neuronal differentiation and survival in-vitro [52]. NSCs derived from knockout mice lacking all three TAM receptors grew slower due to reduced proliferation rates and greater apoptosis, and had reduced neurogenesis rates [52]. Moreover, cultured primary neural stem cells lacking TAMRs have a significant decrease in the expression of nerve growth factor and brain derived neurotrophic factor (BDNF) and a compensational increase in the expression of neurotrophins TrkA, TrkB, TrkC and P75 receptors [52]. These findings suggest a relationship between TAM receptors and neurotrophins to support neural stem cell survival, proliferation and differentiation.
Another developmental role TAMs undertake is the maintenance of neural progenitors. Neural progenitor cells (NPCs) and their neuronal progeny were isolated and purified from developing mouse cerebral cortices (E15.5) and their transcriptome analysis revealed essential roles for TAMRs in maintaining NPCs [53]. All three TAM receptors and PROS1 were enriched in NPCs, and inactivation of both Axl and Mertk promotes cell cycle exit and early differentiation [53].
In the adult brain, newborn neurons can integrate into neural circuitries and allow cellular plasticity. Perturbations in adult neurogenesis are recognized to disrupt hippocampal-dependent learning. As mentioned above, TAMR deficiency impaired adult hippocampal neurogenesis and several studies have been aimed at the role of the TAM ligands in adult neurogenesis. Gely-Pernot reported that inhibiting the bioactivity of both Gas6 and PROS1 by warfarin leads to elevated BrdU incorporation in the SVZ, indicating increased proliferation of NSCs. When an anti-PROS1 function-blocking antibody was perfused into the lateral ventricles, increased NSC proliferation was observed, indicating PROS1 supresses SVZ NSC proliferation [40]. Interestingly, the opposite effect was observed when Gas6-deficient brains were analysed, demonstrating a significant functional variability among the TAM ligands PROS1 and Gas6 in regulating NSC proliferation. This emphasizes that both ligands can differentially induce distinct outcomes in the same cells (Fig. 1). The basis for this functional diversity is not yet understood. Our lab has used genetic tools to inhibit PROS1 expression in a cell-specific manner from NSCs.
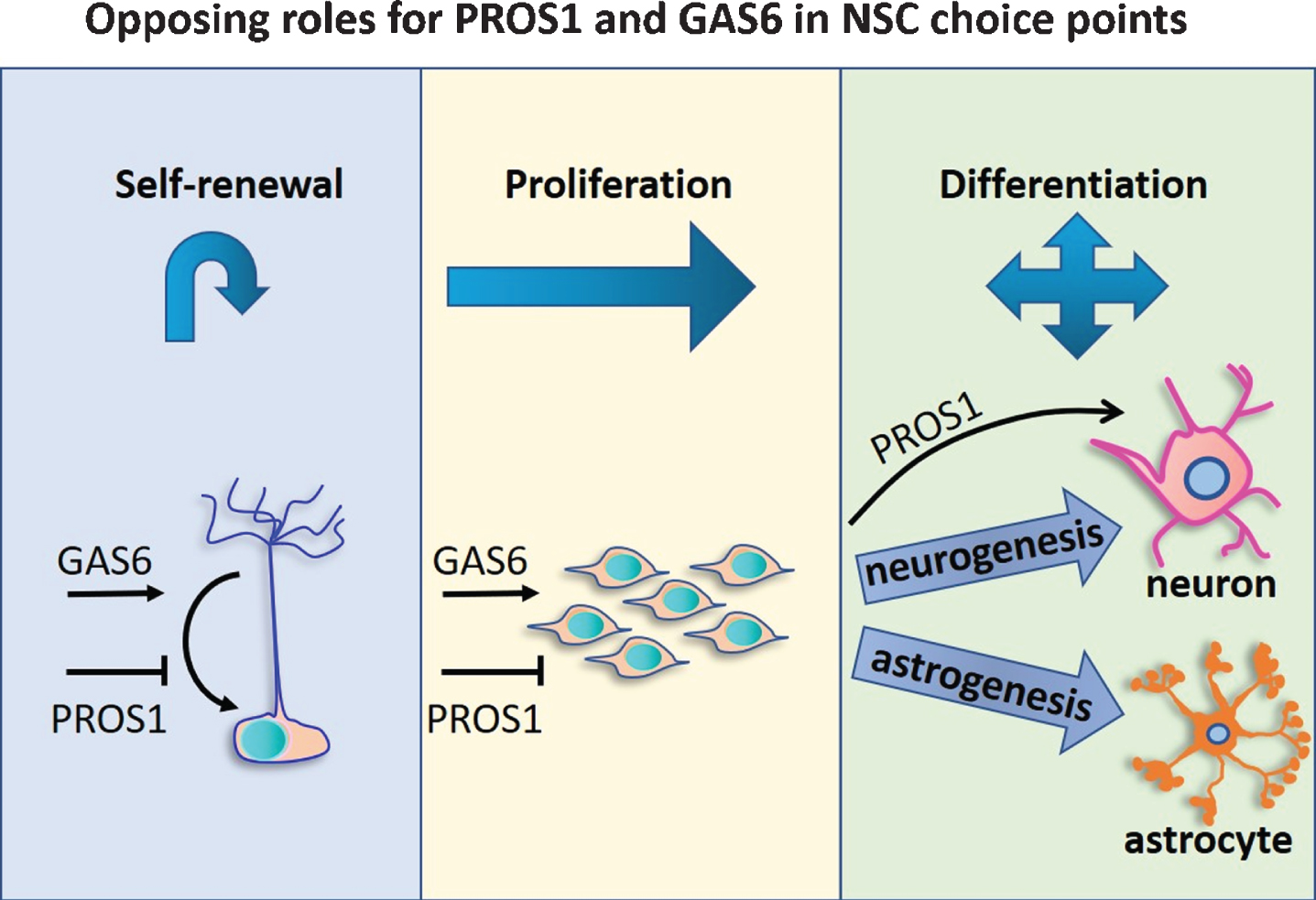
Opposing roles for PROS1 and GAS6 in NSC choice points. Different roles for the TAM agonists PROS1 and Gas6 were revealed through their genetic deletion. Opposing effects are seen on NSC self-renewal, where GAS6 deletion reduces the number of NSCs [40] but PROS1 deletion increased NSC self-renewal [24]. NSC “stemness” is maintained by PROS1, which inhibits NSC proliferation [25], but Gas6 promotes NSC proliferation [40]. PROS1 deletion led to an increase in astrogenesis, indicating its instructive role in the neurogenic fate lineage [25]. By contrast, Gas6 deletion had no effect on the cell fate of differentiated cells [40]. This delicate balance between Gas6 and PROS1 may contribute to NSC homeostasis and neurogenesis.
This approach identified NSC-derived PROS1 acts in an autocrine manner, and affects numerous aspects of both embryonic and adult NSC homeostasis. Analysis of adult Pros1-deficient NSCs (using the Nestin-Cre driver) during embryonic development led to an increase in NSC proliferation and affected the fate of differentiated cells, increasing astrogenesis at the expense of neurogenesis. These findings indicate that PROS1 has an essential instructive role in maintaining NSC quiescence and facilitating neurogenesis [25], in agreement with experiments that neutralized PROS1 protein [40]. We further found that PROS1 promotes NSC quiescence through regulating Notch-1 expression and signaling [25]. A continuing study revealed that lack of PROS1 in NSCs doubled NSC self-renewal, suggesting PROS1 regulates the balance between NSC self-renewal and differentiation. Mechanistically, we found that PROS1 inhibits Bmi-1, which leads to exit from the cell cycle and differentiation [24], summarized in Fig. 2.
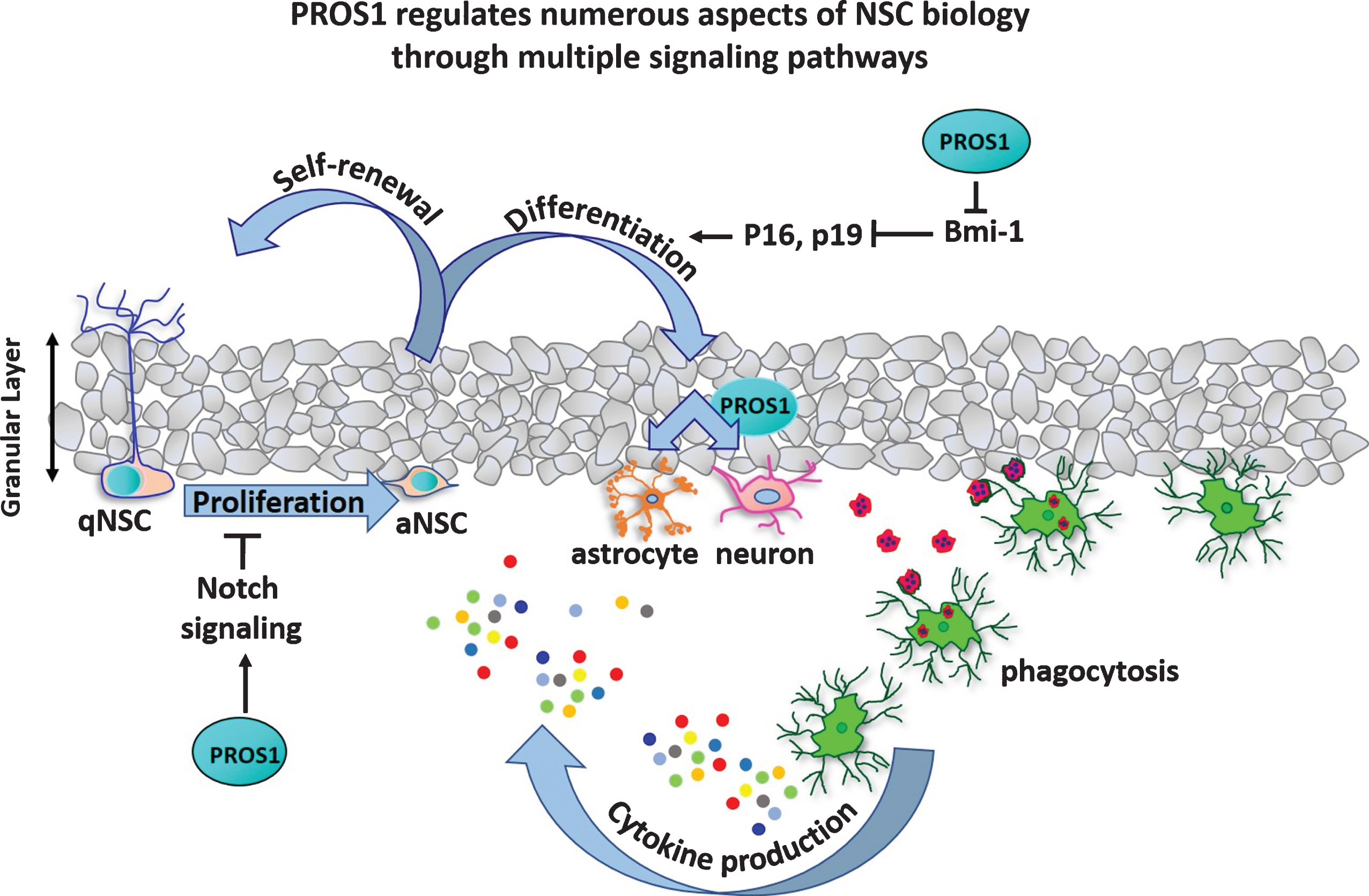
PROS1 regulates numerous aspects of NSC biology through multiple signaling pathways. Schematic of the dentate gyrus granular layer, indicating the influence of PROS1 on different phases of adult NSCs and neurogenesis. Quiescent NSCs (qNSCs) are glia-like and present with a dendritic arbor spanning the granular layer. Quiescent NSCs express PROS1, considered a stem-cell maintenance factor. PROS1 regulates Notch1 signaling, and suppresses NSC proliferation [25]. Upon integration of various signals, qNSCs begin to amplify through proliferation. Amplifying NSCs (aNSCs) also express PROS1, which promotes NSC differentiation by inhibiting Bmi-1, a transcriptional repressor of p16 and p19 which allows for continuation of self-renewing cell divisions [24]. PROS1 expression in differentiated NSCs promotes a neural cell fate over astrogenesis [25]. Within the neurogenic niche, PROS1 is highly expressed by microglia, where it is thought to regulate cytokine expression and phagocytosis of apoptotic neurons, thus influencing neurogenesis in a non-cell autonomous manner.
NSCs and adult neurogenesis are heavily influenced by the neighbouring niche cells, including microglia - the primary immune cells of the CNS [54]. The cytokines secreted by microglia have profound influence on adult neurogenesis [55], and since TAM signaling within immune cells is anti-inflammatory [13, 14, 22], their role in microglia was tested. In line with their immune-suppressive function, cultured TAM-deficient microglia are chronically activated, with hyperactivation of the MAPK/p38/NFκB pathway, and overproduction of pro-inflammatory cytokines, including IL-6, which can be neurotoxic and detrimental for NSC survival and self-renewal [56]. Thus, through modulating inflammatory cytokine signaling within microglia, TAMs can support neural stem cells and adult neurogenesis by restricting the release of toxic inflammatory mediators. Moreover, Gas6 and PROS1 may both modulate adult neurogenesis through regulating cytokine secretion and restrict detrimental immune responses [56, 57]. Such modulation may be utilized as a therapy for diseases characterized by loss of hippocampal neurons or to promote learning and memory through supporting adult neurogenesis.
TAM signaling also supports survival of adult neuronal populations. GAS6 was shown to rescue cortical neurons from amyloid-β induced cell death [58] and mice lacking all three TAMRs had elevated instances of dentate gyrus and cerebellar purkinje cell death [59]. Although this was attributed to a systemic inflammation and impaired BBB integrity [59], the PROS1/Tyro3 axis was shown to provide direct neuronal protection following ischemic brain injury and excitotoxicity [28, 60]. Tyro3 promotes survival of hypothalamic Neuropeptide Y+ neurons [61], as well as the survival of retinal ganglion cells (RGC) and the maintenance of RGC dendrites within the inner plexiform layer of the neural retina. This loss of RGCs resulted in a thinner retina, with reduced photoreceptor and RGC function [62]. While the above examples demonstrate a cell-autonomous protective function for TAM signaling, survival of retinal photoreceptors depends heavily on TAM function in a cell non-autonomous manner. The neighbouring retinal pigment epithelium (RPE) cells are professional phagocytes, which eliminate photoreceptor outer (distal) segments in a circadian fashion [16, 26, 63]. Thus, in different physiological contexts the contribution of TAM signaling to neuronal survival may be either cell-autonomous or non-cell-autonomous.
TAM SIGNALING IN SYNAPTOGENESIS AND SYNAPTIC PRUNING
Synaptogenesis - the establishment of synapses between neurons, occurs robustly during early nervous system development and endures throughout life. After the initial formation of the synapse, the synapse has an ability to strengthen or weaken over time due to fluctuations in activity, commonly known as synaptic plasticity. Underlying these processes is a phenomenon known as long term potentiation (LTP) which strengthens synapses based on patterns of activity and contributes to learning and memory [64]. Tyro3 expression in rats increases in the early post-natal stages correlating with the establishment of synapses [33, 65], and is abundantly expressed in in the CA1 hippocampal field, which is a hot spot for LTP [33, 65]. Tyro3 expression on neuronal processes [61] and its colocalization with post synaptic density protein-95 (PSD-95), a scaffolding protein stabilizing synaptic connections [65] indicates its cell-autonomous role. It was briefly mentioned that Tyro3-/- mice exhibit diminished hippocampal LTP, although the experimental evidence was not presented [15]. In vitro, Gas6 can stimulate Tyro3 phosphorylation in cortical neurons, inducing MAPK and PI3K pathways, vital in the initiation of hippocampal LTP and synapse formation [65]. The specific elimination of weakened and redundant synaptic connections, commonly known as synaptic pruning, is a critical process in the maturation and establishment of proper brain function and strengthening essential synapses. Astrocytes and microglia are the major phagocytes targeting synapses and neuronal debris for elimination. Astrocytic engulfment and removal of synapses were significantly diminished in Mertk-/- mice, leaving redundant excess synapses in the developing and adult neural circuits [37]. Addition of astrocyte-conditioned media significantly increased their phagocytic ability. PROS1 significantly elevated synaptosomes uptake by astrocytes, and since astrocytes express and secrete PROS1 [37], this function is presumably autocrine in nature. Dysfunctional TAM signaling may therefore play a role in aberrant synaptic pruning which is associated with many neuropsychiatric disorders such as schizophrenia [66]. The emerging evidence presented suggests that TAM signaling is involved in both synaptogenesis and the homeostatic regulation of synaptic pruning, supporting brain plasticity and functionality. Yet, the relative and specific roles of the TAMRs and ligands, and the downstream signaling pathways activated in such biological processes are still unknown.
TAM SIGNALING IN THE NEUROIMMUNE INTERFACE
TAMR signaling in immune cells is anti-inflammatory. The nervous system is separated from the peripheral immune system by the blood-brain barrier (BBB), a semipermeable neurovascular unit essential to keep brain homeostasis. In the CNS microglia are the principal resident innate immune cells accounting for 10–15% of glial cells found in the brain [67]. Belonging to the myeloid lineage, microglia originate in the embryonic yolk sac and migrate to colonize the mouse brain from embryonic day 9.5 [68, 69]. Microglia are essential in regulating neural development and maintaining homeostatic conditions, preserving neuronal networks and are the primary immune defence to challenges faced by the CNS [70, 71]. Microglia are primarily regarded for their phagocytic and immune- inflammatory functions in response to foreign material, damaged or apoptotic cells and secretion of trophic factors which can instigate repair mechanisms. Aberrations to microglia functionality can contribute and lead to pathological processes associated with many diseases and has been linked to neurodegeneration [72, 73]
As potent phagocytes, microglia prevent the accumulation of toxic products leading to autoimmunity [74, 75]. Moreover, microglial phagocytosis is important for synaptic regulation (as mentioned above), myelination and repair mechanisms, shown to maintain a healthy functional brain tissue and prevent neuroinflammation and neurodegeneration. Various molecular mechanisms participate in the different microglial functions, with the TAM receptors and ligands being key. Although TAM receptors share functional homology, they are activated under different conditions: Mertk is the dominant receptor in resting microglial functions, while Axl functions in acute inflammatory environments [36, 76]. In Axl-/-Mertk-/- double knockout mice phagocytosis is impaired with subsequent accumulation of apoptotic cells [36]. Axl is important for the migration of microglia (and infiltrating macrophages) to damaged tissue and the clearance of debris [77]. Moreover, without TAMs microglia produce excess proinflammatory cytokines harming homeostasis and repair mechanisms [56, 57]. Gas6 was shown to inhibit TNFα production in microglia [78], however, the relative roles of PROS1 and GAS6 in microglial biology have not been investigated.
In addition to serving as TAM agonists, PROS1 and Gas6 are necessary for TAM function, by providing a bridging moiety linking the apoptotic cell (or the cell fragment to be uptaken) and the phagocyte. An N-terminus Gla domain of approximately 60 amino acid binds externalized phosphatidylserine, an “eat me” signal exposed by apoptotic cells, in a Ca2+-dependent manner [79–81]. This bridging capacity drives immune cells including microglia to remove apoptotic cells via phagocytosis, and instigates anti-inflammatory signaling at the same time [30].
TAM SIGNALING IN INFLAMMATION AND DISEASE
The TAM pathway is key in regulating immune homeostasis though mediating anti-inflammatory responses [1, 8, 59]. Inflammation is a complex biological response to perturbations such as pathogens, toxins and trauma, and being primarily a defensive reaction involves immune cells and complex molecular signaling [82]. However, the magnitude of inflammation must be carefully controlled. Elevated and sustained (chronic) inflammation in the CNS are stress inducers promoting ‘secondary injury’ and hindering repair and remyelination [82], further exacerbating damage. As TAMs are involved in phagocytosis, debridement and modulation of inflammation, it is no surprise that their dysfunction is involved in autoimmune, inflammatory and neurodegenerative diseases.
The dysregulation of TAM signaling has been implicated in pathological processes leading to neuroinflammation, myelination abnormalities, neurodegeneration and ischemic injury to neurons (Fig. 3). Mice that were genetically deleted for all three TAM receptors had systemic chronic inflammation and autoimmunity, which lead to brain damage, BBB breakdown, release of pro-inflammatory cytokines, protein aggregate deposition and neuronal death [59]. Given the cellular tasks performed by TAMs are anti-inflammatory in nature, their therapeutic potential in autoimmune disease has been investigated. Multiple sclerosis (MS) - a complex autoimmune disease involving many, if not all TAM-related aspects is one such disease, discussed below.
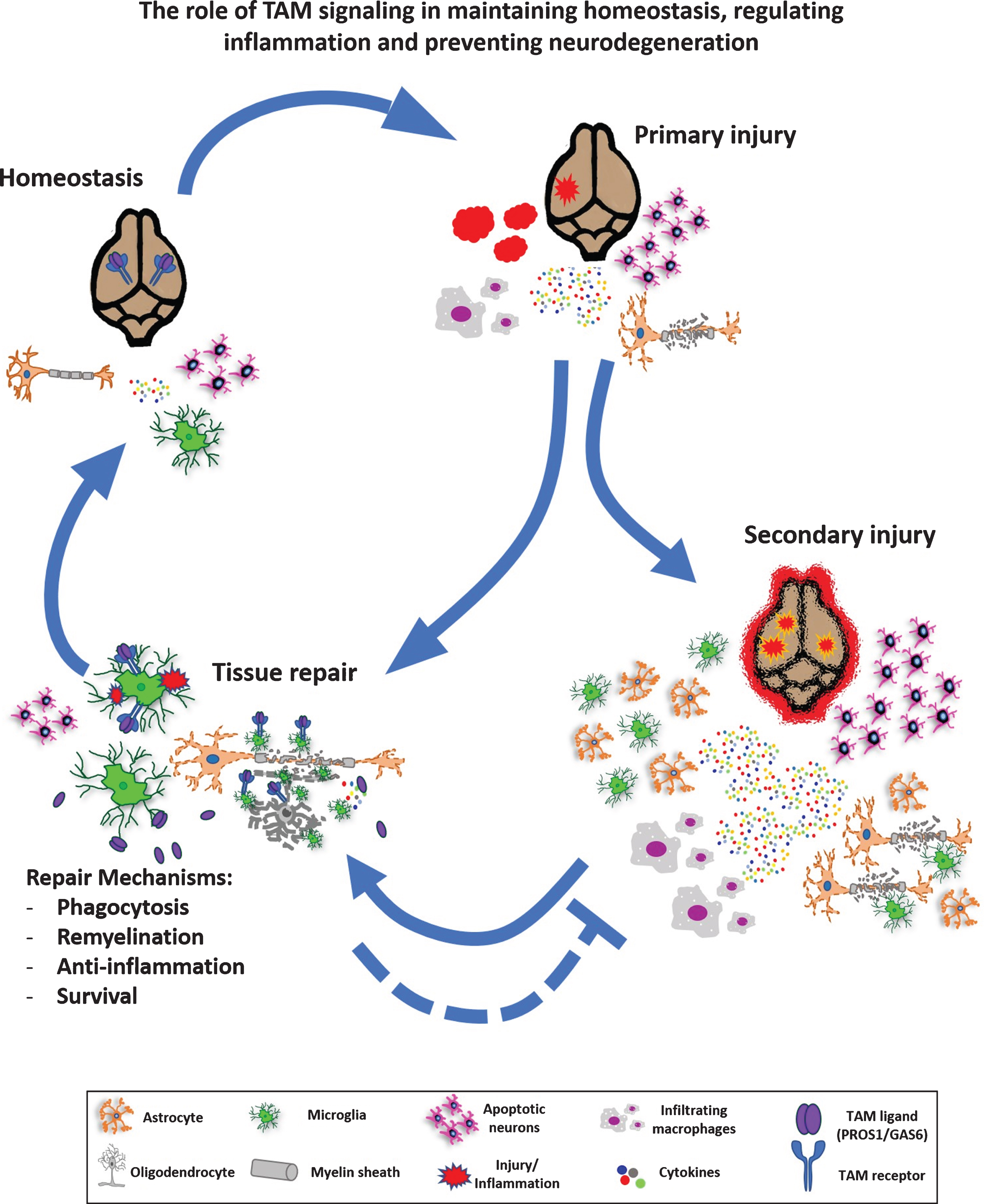
The role of TAM signaling in maintaining homeostasis, regulating inflammation and preventing neurodegeneration. At steady state, the TAM pathway mediates homeostasis via regulation of NSCs, microglial activity, myelination and phagocytosis. Upon injury, such as acute trauma, a demyelination event or neurodegenerative conditions, inflammation is instigated which may induce neurogenesis, elevated cytokine secretion, microglial and astrocytic gliosis, oligodendrocyte damage and demyelination. If unresolved, primary injury further develops into secondary injury with high toxicity, necrosis and may potentially develop into a chronic inflammatory condition, with activation of microglia and astrocytes, demyelination and toxin release before a new homeostasis is reached with accompanying tissue repair. Alternatively, primary injury may be resolved without secondary injury. TAM signaling is involved in various aspects of homeostasis and tissue repair, as detailed in the main text.
TAM SIGNALING IN MULTIPLE SCLEROSIS
Multiple sclerosis is a complex disease, involving inflammation, demyelination/remyelination dynamics and axonal damage. Oligodendrocytes (OD) and Schwann cells are responsible for myelination in the CNS and PNS respectively, protecting and insulating axons, necessary for their proper function. Recent studies indicate that in addition to anti-inflammatory and neuroprotective functions, TAM signaling also promotes myelination [83, 84].
The pathogenesis of MS remains largely unknown but the processes of demyelination and remyelination provide sound insight into dysfunctional pathways that may underlie MS and impair neurological function leading to physical, mental and occasionally psychiatric symptoms [85]. Inflammation in MS results from immune cell infiltration, microglia activation and tissue damage, also affecting OD viability and myelin maintenance. The diversity of MS pathology is further complicated by acute and chronic courses, eventually leading to lesions. The two major mouse models used to study the TAM pathway in MS are the Experimental Autoimmune Encephalitis (EAE) model, induced by immunization with myelin oligodendrocyte glycoprotein (MOG) and the cuprizone-induced demyelination/ remyelination model [86]. The former includes significant involvement of immune cells including CD4+ and CD8+ T cells, followed by monocytes/macrophages infiltration, which then elicit microglia and astrocyte reactivity [86]. Administration of cuprizone - a copper chelator - in the latter model targets ODs, causing their degeneration, and subsequent demyelination [86]. When cuprizone is withdrawn from the diet, remyelination occurs. Thus, whereas immune cell infiltration has been reported for both models, EAE includes both demyelination and stronger inflammatory involvement, while the cuprizone model is primarily a demyelination and remyelination model, also involving OD differentiation and maturation [86].
The TAM receptors and ligands have all been associated with these different aspects of MS biology both in humans and in experimental MS models in mice [77, 84, 87–92]. Mertk and Axl are upregulated in human chronic MS lesions and genome wide studies showed that Mertk is a risk gene for MS susceptibility [88, 91]. Gas6 was upregulated in patient’s cerebrospinal fluid (CSF) compared to controls and plasma levels of total but not free PROS1 was reduced in MS patients compared to controls [89, 90].
The degraded myelin and cell debris, overactivation of immune cells and hyperinflammation in the CNS and oligodendrocyte survival and function all inhibit tissue repair mechanisms, successful remyelination and consequent recovery [93, 94]. TAMs, chiefly Mertk, mediate the uptake of apoptotic cells and myelin debris by recruiting microglia and macrophages in mouse models and in human cells to the injury site - a process essential for debridement and allowing the myelin sheath to repair [39, 93, 94]. The upregulation of Axl in disease states is important for the engagement of microglia and infiltrating macrophages in clearing apoptotic cells and debris, necessary to alleviate disease progression [77]. Mertk and Axl pathways also mediate myelin clearance by phagocytosis in Schwann cells of the PNS and Mertk in myeloid cells [39, 95]. Moreover, since phosphatidylserine is present in myelin [96], it may be recognized by PROS1 and Gas6 for debridement. A recent work identified Mertk in microglia activation and myelin clearance following cuprizone-induced demyelination. Moreover, remyelination in Mertk-deficient mice was impaired following cuprizone withdrawal [97]. Mertk-/- microglia showed an elevated Interferon gamma (INFγ) signature, which impeded on their normal function [97]. Whether PROS1, GAS6, or both activate Mertk in these contexts remains to be determined. Chronically inflamed tissue, as in MS, can cause further deterioration due to glial cell activation accompanied by proinflammatory cytokine production, further exacerbating demyelination and axonal nerve damage [75]. Loss of Axl itself or its ligand Gas6 leads to inflammation, impaired clearance of myelin debris and diminished remyelination [77], pointing to their functional importance in minimizing MS pathology.
Tyro3 has been implicated in developmental myelination of the optic nerve and the absence of Tyro3 accelerated demyelination which resulted in thinner corpus callosum myelin sheaths [98]. Interestingly, loss of Tyro3 did not affect the density of microglia or OD progenitor cell recovery after cuprizone treatment, indicating the survival of these cells is not affected by loss of Tyro3 function [99].
Based on the above, stimulating TAM functions by their ligands Gas6 and PROS1 is hypothesized to benefit multiple MS-related aspects leading to better patient outcomes. Indeed, Gas6 delivery stimulates remyelination both in vitro and in vivo and Gas6/TAM signaling favours myelin repair and recovery from damage [83, 90, 100]. However, the effects of TAM inhibition on myelination are only partially phenocopied by the loss of Gas6 implying a potential compensatory role for PROS1 [98]. Furthermore, the recent discovery that PROS1 both induces phagocytosis and inhibits inflammation in macrophages in vitro and in vivo [22, 101] suggests it may support a similar role in microglia following myelin and cellular damage. Taken together, activation of Axl and Mertk by PROS1 and Gas6 is postulated to re-establish homeostasis by regulating inflammation, promoting debris clearance and directly supporting oligodendrocyte cell survival. These functions not only support the effector cells, but also promote an environment that is favourable for remyelination. Given ODs, astrocytes and microglia become reactive in MS lesions, the roles of TAM signaling molecules in these cells should also be clarified in the context of MS.
TAM SIGNALING IN NERVOUS SYSTEM INJURY AND NEURODEGENERATION
Insults to the CNS and PNS share common pathological mechanisms, but specific reactions are also characteristic of the particular insults. Traumatic brain injury, stroke, hypoxic or ischemic injury and nerve crush can all result in inflammation, a milieu of cell death and neurological damage, and TAM signaling molecules play roles in the recovery process. Gas6 and PROS1 both support neuronal survival under stress conditions [28, 102]. Gas6 functions as a neurotrophic factor also exhibiting anti-apoptotic effects in serum-starved cultured hippocampal neurons [102]. PROS1/Tyro3 signaling protects from N-methyl-D-Aspartate receptor (NMDA-R) mediated neurotoxicity, and from hypoxic/ischemic injury [28]. PROS1 was also identified as a neuroprotectant during ischemic brain injury, as treatment with PROS1 resulted in decreased hypoxia-induced damage and reduced infarction and edema [60]. PROS1 is upregulated in the rat sciatic nerve as early as 1-2 days following nerve injury, suggesting its function in the immediate response to injury [5]. A recent study focusing on the peripheral nervous system identified that TAMs mediate phagocytic uptake of myelin following nerve injury by Schwann cells [95]. Moreover, TAM-deficient microglia display reduced process motility and delayed convergence to sites of injury, limiting repair mechanisms [36].
Neurodegenerative diseases are also accompanied by chronic neuroinflammation, with microglia and astrocytes as key mediators [103]. Neurodegenerative diseases are commonly recognized by the accumulation of pathological protein deposits such as alpha synuclein in Parkinson’s disease (PD) and beta- amyloid and tau in Alzheimer’s disease (AD) [104]. These protein deposits can cause inflammatory immune reactions characterized by the release of pro-inflammatory cytokines and reactive oxygen species which lead to progressive neurodegeneration, brain atrophy and associated mental and physical deficits [72].
AD is the largest cause of dementia, causing 60–70% of cases worldwide [105]. Although the pathophysiology of AD is not completely understood, increased prevalence of AD is seen among patients who experienced traumatic brain injury, suggesting AD is linked to CNS injuries and inflammation [106, 107]. The presence of beta amyloid plaques and tau tangles typical to AD is thought to cause neuronal death and subsequent brain atrophy [72]. TAM signaling is understudied in the field of AD, however, a role for Tyro3 in AD has been indicated. The overexpression of Tyro3 in the hippocampus led to a reduction in amyloid beta plaques, while the knockdown of Tyro3 in a transgenic AD mouse model led to more amyloid beta plaques [108]. Within microglia, Axl was identified to downregulate inflammation and improve microglial phagocytosis via PPARγ/HSP90β both in vitro and in a mouse model of AD, also measurable by cognitive tests performed in these mice [109]. This study investigated the effect of Jujuboside A (JuA) on early-onset Alzheimer’s disease. JuA is an antioxidant possessing neuroprotective and anti-inflammatory proterties. JuA recently shown to promote the clearance of Amyloid-β1-42 (Aβ42), which is associated with a higher risk for developing AD, through Axl-mediated pathways [109]. A recent study found that in-vivo clearance of Aβ plaques is significantly inhibited in Axl-/-Mertk-/- mice, along with accumulated debris in line with their role in apoptotic cell clearance. Surprisingly, double knockout mice had fewer Aβ dense-core plaques than WT mice, leading to the conclusion that amyloid dense-core plaque deposition is actually supported by TAM-mediated phagocytosis [110] - a somewhat contradictory finding which calls for further research. In humans, research has been limited but Axl was found to be one of several CSF protein biomarkers preceding a reduction in Aβ42 [111]. Another recent study has found the upregulation of PROS1 and Gas6 in the frontal cortex of AD patients [112]. Yet another study found elevated Gas6 levels in CSF of AD patients compared to controls which correlated with improved cognitive scores suggesting Gas6 could play a defensive role in AD progression [113].
PD is characterized by motor and non-motor symptoms, commonly recognized by a resting tremor, and causes stiffness and progressive slowing of movement. The aggregation of α-synuclein found in Lewy bodies in the CNS and PNS is a central pathology to the disease [114]. The pathophysiology of PD is still largely a mystery and the role of TAM signaling in PD is not yet completely understood. In the SNCAA53T mouse model of PD overexpressing the human α-synuclein with the PD-associated A53T mutation in neurons, both Axl and its soluble form (sAxl)- an inflammatory biomarker- were upregulated in microglia of diseased spinal cords [36]. When SNCAA53T mice were crossed to Axl-/-Mertk-/- mice, the survival of the mutated mice slightly extended. It is not yet understood whether lack of Axl and Mertk in the SNCAA53T model influences inflammation, phagocytosis, or both. Another aspect of Axl and Mertk in PD is their transcriptional regulation by Nrf2, and potentiating their phagocytic and immune regulatory functions [115]. In PD, Nrf2 deficiency is associated with protein aggregation, inflammation and neuronal death. suggesting dysfunctional Axl and Mertk may play a role in PD [115]. A recent report describing exposure of phosphatidylserine by live neurons expressing Tau filaments, showed that phosphatidylserine exposure mediates their phagocytic uptake by microglia through milk fat globule-EGF factor 8 (MFGE8) and nitric oxide (NO) [116]. In light of this finding, TAM-mediated phagocytosis may actively modulate the development and progression of neurodegenerative diseases with pathological Tau aggregation, including PD.
The contribution of microglia has been associated both positively and negatively with neurodegeneration. Microglia activation can be beneficial and protective helping clear the protein deposits aggregating in these disorders such as MS, AD and PD [103]. On the contrary, microglia dysfunction often occurs early on in disease compromising the clearance of proteins and accruing to the inflammatory response [103]. Further research is necessary to determine the role of TAM signaling in neurodegenerative disease and directing therapeutics from a neuroinflammatory towards a neuroprotective and reparative response.
CONCLUSION
TAMs and their ligands have multiple and diverse physiological roles in many biological systems, including the nervous system. Throughout the nervous system TAMRs, PROS1 and Gas6 are widely distributed across many tissue and cell types and often co-expressed allowing functional diversity. From development throughout adulthood TAM signaling partakes in many important roles detailed in this review. TAM signaling dysfunction can cause disease, with a direct impact on inflammation due to their key role regulating inflammation, or as a secondary outcome, due to their role in phagocytosis, in which deficiencies can lead to an accumulation of apoptotic cells. The association between neuroinflammation and neurodegeneration is advancing rapidly with a novel therapeutic focus to downregulate and prevent secondary inflammation and shift to a neuroprotective response. The modulation of TAM signaling has great potential for developing new therapeutic strategies across a wide spectrum of pathologies and diseases due to their distinct and overlapping networks. Therapies effectively modulating TAMs could stimulate phagocytosis and rapid debridement of damaged cells and debris, remyelination, neurogenesis and shift the inflammatory response to be neuroprotective. However, development of an effective therapeutic strategy to modulate TAMs depends on the complete understanding of the interactions between TAMs and their ligands in different cell types. Future research lies in attaining deeper knowledge of TAM signaling and thorough investigation of the possible side effects of modifying specific pathways.
Footnotes
ACKNOWLEDGMENTS
We apologize to our colleagues if their original studies are not mentioned owing to space limitations.
FUNDING
This review was supported by the Israel Science Foundation Grant 655/18 to T. Burstyn-Cohen.
DECLARATION OF CONFLICTING INTERESTS
The authors declare no potential conflicts of interest with respect to finances, authorship or otherwise related to the publication of this article.