
Abstract
Adult neurogenesis in the mammalian brain, including in humans, occurs throughout life in distinct brain regions. Alterations in adult neurogenesis is a common phenomenon in several different neurodegenerative disorders, which is likely to contribute to the pathophysiology of these disorders. This review summarizes novel concepts related to the interplay between autophagy and microRNAs in control of adult neurogenesis, with a specific focus on its relevance to neurodegenerative diseases.
INTRODUCTION
Prior to the 1990’s, the generation of new functional neurons from progenitor cells was thought to be restricted to development. Despite the fact that ongoing neurogenesis in the adult rat brain was suggested by Altman et al. [1, 2] and Kaplan et al. [3] in the 1960’s and 1970’s, respectively, it was not until the end of the twentieth century, with the introduction of bromodeoxyuridine (BrdU), that it was generally accepted that new neurons are generated throughout life in the mammalian brain [4–7].
It is now well established that adult neurogenesis occurs in most mammals. In the human brain this phenomenon is debated but it appears to occur primarily in the hippocampus and in the striatum [8–10]. In the hippocampus, adult neurogenesis is thought to contribute to pattern separation, while the exact function of the adult-born striatal neurons is currently unknown [11–14]. Adult neurogenesis can be modulated by several external stimuli, as well as disease, including neurodegenerative disorders [15–17]. For example, adult-born striatal neurons are selectively degraded in Huntington’s disease (HD) [18–20]. Thus, a better understanding of the molecular regulation of adult neurogenesis in both the normal and diseased brain may lead to the development of novel therapeutic strategies. These strategies may be used to modulate human adult neurogenesis and, ultimately, treat diseases where adult neurogenesis is impaired such as HD.
In this review, we will discuss a novel role for microRNAs (miRNAs), in the regulation of transcriptional networks in adult neurogenesis through the regulation of autophagy. Furthermore, we consider the relevance of this process in relation to the impairment of adult neurogenesis found in neurodegenerative disorders.
ADULT NEUROGENESIS
In most mammalian brains, adult neurogenesis occurs continuously and spontaneously in two distinct areas, the so-called stem cell niches: in the subgranular zone (SGZ) in the dentate gyrus of the hippocampus [21], and in the subventricular zone (SVZ) in the lateral ventricles [22] (Fig. 1A-B). Neural stem cells (NSCs) reside in both areas throughout life and give rise to new neurons. In addition, there have been several reports indicating the presence of NSCs and spontaneous adult mammalian neurogenesis in other brain regions such as the striatum, spinal cord, the cerebral cortex or the retina [18, 23–25].
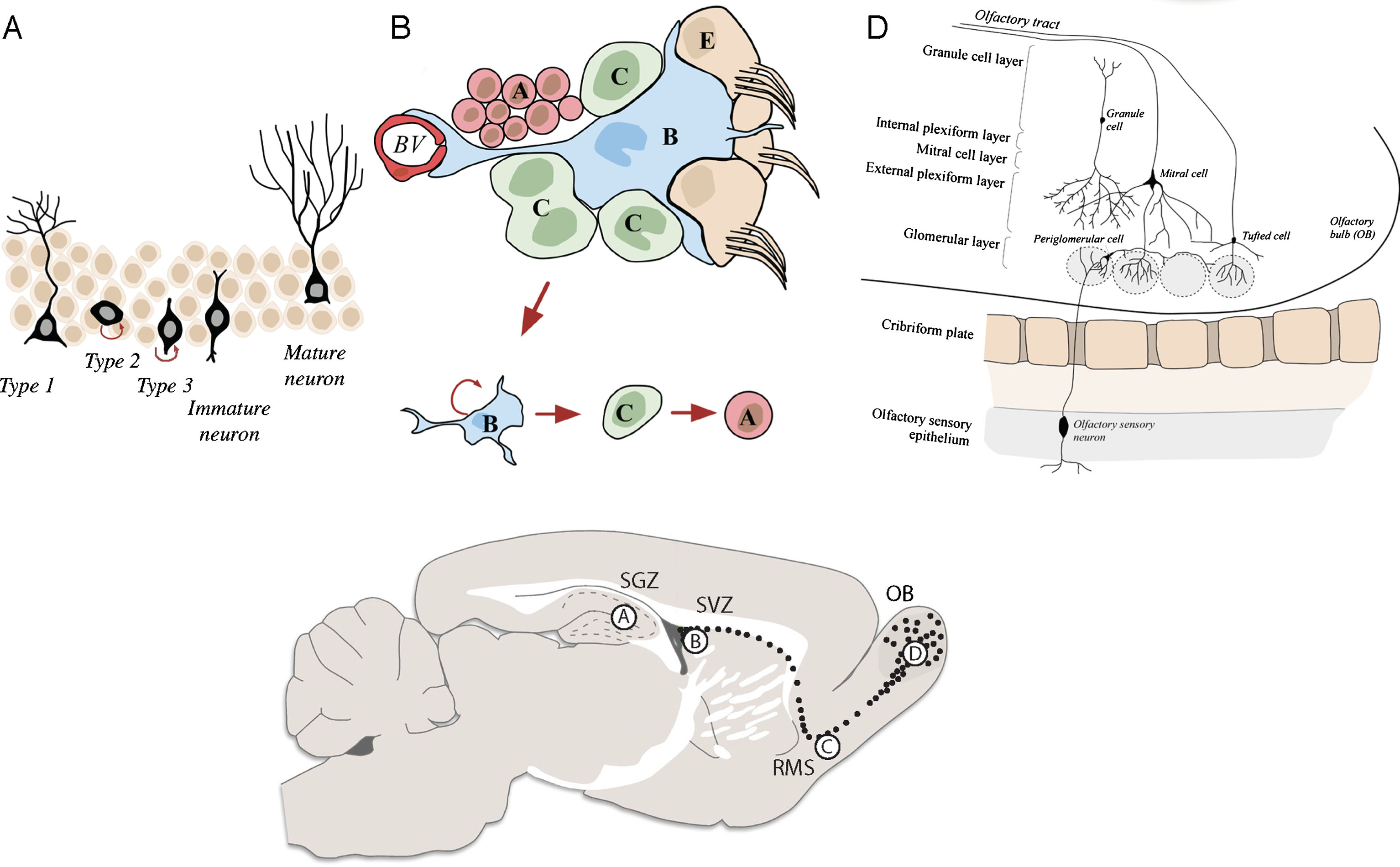
Schematics of the neurogenic niches in the adult mouse brain. A) The SGZ is located in the dentate gyrus of the hippocampus. Radial astrocyte-like NSCs (type 1 cells) give rise to intermediate progenitor cells (type 2 cells), which in turn give rise to neuroblasts (type 3 cells) that generate mature granule cells. These granule cells subsequently integrate into the granule cell layer of the dentate gyrus. B) The SVZ is located in the lateral ventricles of the adult mouse brain. Neural stem cells (called type-B cells) generate transient amplifying progenitors (type C cells), which give rise to neuroblasts (type A cells) that migrate along the RMS (C) into the OB (D).
In the rodent SVZ, radial glia-derived astrocyte-like cells (type B cells) function as NSCs (Fig. 1B). These cells give rise to rapidly dividing progenitors (type C cells), which form neuroblasts (type A cells) [26]. These neuroblasts tangentially migrate along the rostral migratory stream (RMS) (Fig. 1C), into the olfactory bulb (OB), where they radially migrate from the OB centre to the periphery in order to reach distinct OB layers. There, they differentiate to mature interneurons and integrate into the pre-existing neuronal circuitry forming synaptic connections with OB projection neurons [22] (Fig. 1D). Interneurons generated during adult neurogenesis are classified as either granule cells (GC, 95% of all OB interneurons) or periglomerular cells (PGCs). The majority of these cells are GABAergic (95%, GC and PGCs) with only a small percentage being dopaminergic cells (PGCs). Importantly, despite thousands of new interneurons reaching the OB every day, only half of them survive and are able to integrate [22, 27]. The remaining cells undergo programmed cell death (PCD) by apoptosis, necrosis or autophagic cell death [28].
The analysis of human adult neurogenesis in more detail was recently revolutionised by the Frisén-lab through the development of assays to measure labelled nucleotide incorporation [8]. The level of the isotope carbon-14 following cold war atomic weapons testing can be used to determine when different cell populations were generated providing evidence for robust neurogenesis in the human adult hippocampus and striatum [18, 29]. The origin of new adult-born striatal interneurons is not known, but it has been speculated that they derive from either the SVZ or from local neuronal progenitors in the striatum itself [18, 30]. The morphology and functionality of these adult-born striatal neurons is similar to the postnatally generated interneurons in the rodent OB and dentate gyrus, with neurons having a single primary dendrite [31–34].
MICRORNAS AND ADULT NEUROGENESIS
Adult neurogenesis is a remarkably regulated process including a precise temporal control of migration, differentiation, integration and maturation of new-born neurons. At a molecular level, miRNAs are attractive candidates for regulating these processes since they have the potential to control large transcriptional networks. Indeed, several miRNAs have already been implicated in adult neurogenesis in both the SGZ and the SVZ: miR-124, for instance, a neuron-specific miRNA, which expression starts in neural progenitor cells (NPCs), and increases during neuronal maturation, serves as a key neuronal determinant in the mouse SVZ [35, 36]. Other examples for miRNAs regulating adult neurogenesis are miR-9, which serves as a negative regulator providing a balance between NSC proliferation and differentiation [37] and miR-137, which is part of a crosstalk with epigenetic regulators and modulates neuronal maturation [38, 39]. Let-7, which will be discussed in more detail later in this review, was identified as the most abundant miRNA in newborn adult OB interneurons and to be crucial for their functional radial migration and maturation [40]. Another important miRNA in adult-born OB interneurons is miR-125b, which controls functional integration of this cell population into the OB [41].
MiRNAs are small, single-stranded, non-coding RNAs with an average size of 20–24 nucleotides (nt). At present, more than 2500 mature miRNAs are annotated in the human genome [42, 43]. While most miRNAs are highly conserved among species, some miRNAs have been found to be specifically expressed in primates and humans including several that are enriched in the brain [44, 45]. It is estimated that approximately 60% of all protein-coding genes in the human genome undergo miRNA regulation. Given that one miRNA can target hundreds of genes in a cell, and one gene can have several miRNA target sites, it is reasonable to say that miRNAs act as regulators of complex genetic networks [46–50].
The transcription of miRNAs typically starts with the generation of a long primary miRNA transcript (pri-miRNA; >1kb), which is often generated by Polymerase II (Pol II) (Fig. 2A). These long transcripts either give rise to individual miRNAs, or to miRNA clusters which form a characteristic stem-loop structure [51, 52]. While still in the nucleus, the pri-miRNA transcripts are recognised by the microprocessor complex (Fig. 2B). This complex consists of Drosha, an RNAse III enzyme, and two RNA binding proteins called DiGeorge Critical Region 8 (DGCR8). The microprocessor complex cleaves the pri-miRNA into an approximately 60–70 nt long hairpin-structured precursor miRNA (pre-miRNA) [53–55]. The pre-miRNA is subsequently exported into the cytoplasm by Exportin-5 [56, 57] (Fig. 2C), where the Drosha-induced overhang is recognised by another RNAse III enzyme called Dicer (Fig. 2D). Together with the co-factor, Transactivation-responsive RNA-binding protein (TRBP), Dicer cleaves the pre-miRNA to form mature miRNA duplexes. The miRNA duplexes are subsequently transferred onto Argonaute (AGO) proteins. Once transferred, the miRNA strands are unwound with one of the strands being incorporated into the RNA-induced Silencing Complex (RISC), and the other strand disassociating and degrading [58] (Fig. 2E). The incorporated miRNA strand subsequently guides the RISC to mRNAs, to which it preferentially binds in the 3’UTR [59] resulting in a combination of translational repression, deadenylation and decay.
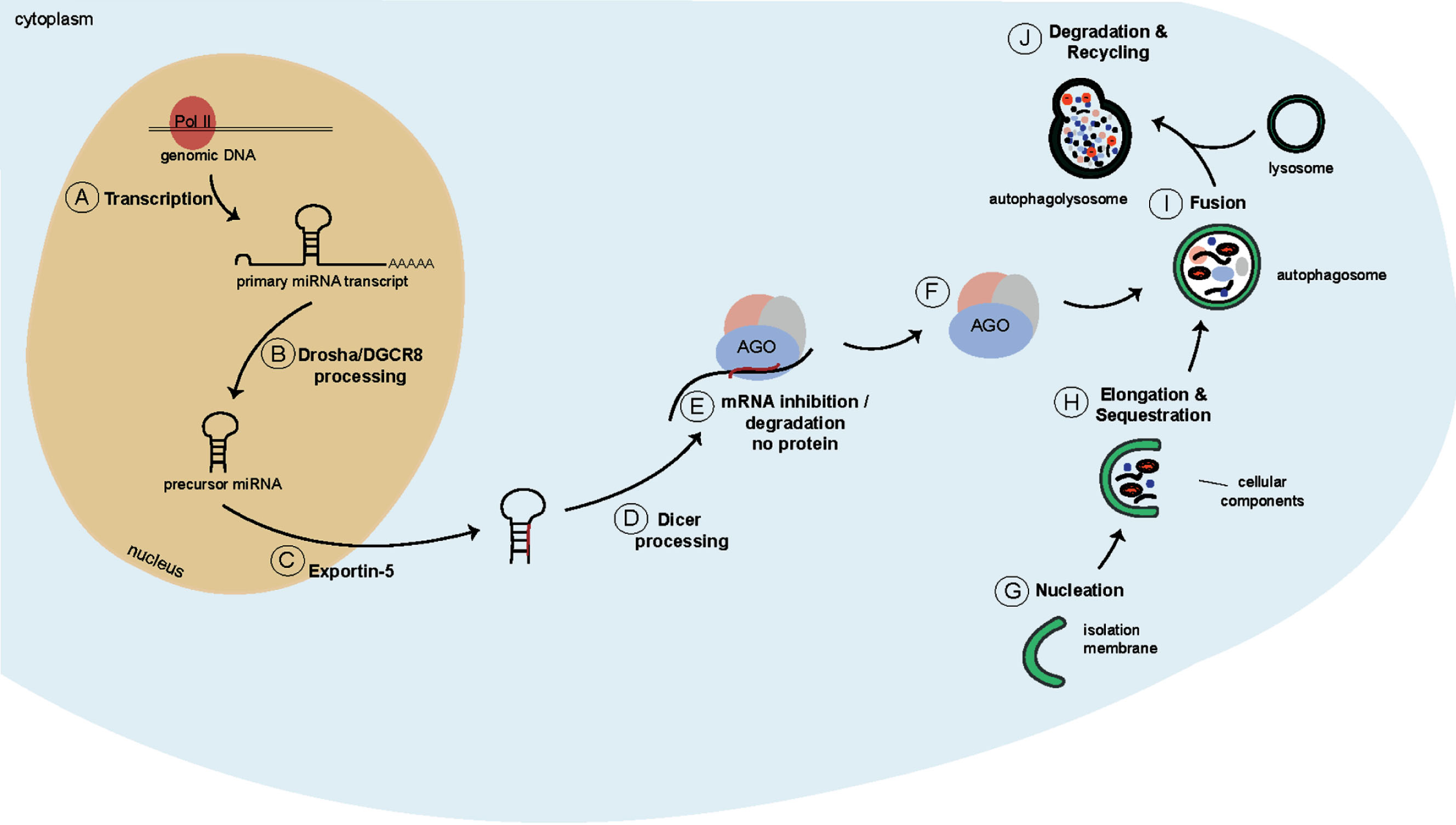
Schematics of the miRNA biogenesis pathway and of macroautophagy. A) A long primary miRNA transcript is generated by Pol II. B) This transcript is recognised by the microprocessor, and is cleaved into a hairpin-structured precursor miRNA. C) The precursor miRNA is transported into the cytoplasm by Exportin-5. D) Dicer cleaves the precursor miRNA into mature miRNA duplexes. E) One strand is bound by AGO proteins and targets mRNAs to induce a combination of translational repression, deadenylation and decay. F) Dicer and AGO2 containing complexes are targeted for degradation by autophagy. G) During autophagy first, an isolation membrane is formed to engulf cytoplasmic cargo (H) thereby giving rise to a structure called autophagosome (I). J) The autophagosome subsequently fuses with a lysosome to form the autophagolysosome where the cargo is degraded.
Interestingly, the homeostasis of the miRNA machinery has recently been suggested to be regulated by autophagy since miRNA-free AGO2 and Dicer are selectively degraded by autophagy (Fig. 2F). This might serve as an important checkpoint for miRNA biogenesis, given that the accumulation of empty Dicer and AGO2 might interfere and compete with loaded proteins [60–62]. In addition, several miRNAs have been linked to the regulation of neuronal autophagy, including miR-34 that controls the neuronal differentiation of NSCs, which regulation is at least partially dependent on autophagy [63]. MiR-299 modulates neuronal survival programs by suppressing autophagy-related gene 5 (ATG5) and antagonizing caspase-dependent apoptosis [64]. MiR-137, which has been known to be involved in neuronal maturation and neurogenesis, also regulates starvation induced autophagy by targeting ATG7 [38, 39, 65]. Let-7 is highly expressed in the brain and its expression peaks during neurogenesis and neural differentiation. Let-7 controls several genes in a pathway upstream of mammalian target of rapamycin (mTOR), called the amino acid sensing pathway, thereby positively regulating autophagy in primary neurons [66, 67]. MiR-124 role has been extensively studied in the brain and has been connected to autophagy by regulating the 5’ adenosine monophosphate-activated protein kinase (AMPK)/mTOR pathway [35, 36, 68–71]. Together, these data suggest that there is a crosstalk between miRNAs and the regulation of autophagy in the brain.
AUTOPHAGY AND ADULT NEUROGENESIS
Macroautophagy, hereafter referred to as autophagy, is an evolutionary conserved lysosomal degradation pathway that allows degradation and recycling of cellular components [72]. During autophagy a double-membraned vesicle, called an autophagosome, is generated to engulf cytoplasmic cargo (Fig. 2G, H). The autophagosome subsequently fuses with a lysosome (Fig. 2I), giving rise to an autophagolysosome, which is where the degradation of the cytoplasmic cargo takes place [72, 73] (Fig. 2J). Autophagy allows cells to react to changing nutrient conditions and to maintain cellular homeostasis [74].
In the brain, autophagy is active at basal levels in almost all cells [75]. Since neurons are post-mitotic and have large and extensive cytoplasmic compartments due to dendrites and axons, they are particularly dependent on efficient clearing of organelles and cellular waste. In line with this, impaired autophagy is often found in neurodegenerative disorders [74, 75]. The importance of autophagy in the central nervous system (CNS) has, for example, been demonstrated by several reports implicating autophagy in embryonic and adult neurogenesis, and there are several studies that link autophagy-related genes to adult neurogenesis [76–79]. While these studies indicate that autophagy is important in adult neurogenesis, the molecular mechanisms that regulate autophagy during adult neurogenesis are still unknown. Given the important role of miRNAs in the control of both autophagy and adult neurogenesis, we recently explored a link between these two processes by investigating the role of the miRNA let-7 [40].
LET-7
Let-7 was one of the first miRNAs to be identified and was initially found to be important for developmental timing in C. elegans [80]. The let-7 family is highly conserved over species. In the mouse and human genome, it is comprised of 9 mature miRNAs encoded from 12 different genomic loci, of which some are part of miRNA clusters [81]. Let-7 is highly expressed in the brain and regulates proliferation, differentiation and cell cycle progression of NSCs [67, 82]. Furthermore, let-7 was demonstrated to regulate the cell fate of neural progenitor cells (NPCs) by targeting the chromatin-associated protein HMGA2 [83]. As mentioned above, a recent study also found that in neurons let-7 targets several genes in the amino acid sensing pathway, thereby positively regulating autophagy [66].
We recently found that let-7 is the most abundant miRNA family in new-born interneurons in the OB, by conducting knock-down studies of let-7, by injecting a let-7 sponge construct (LV.let-7.sp) into the RMS, we found that while cells which lack let-7 could reach the centre of the OB, they failed to radially migrate into its outer layers [40]. In addition, morphological analysis revealed that knock-down of let-7 resulted in decreased neurite length and a reduced number of branch points, suggesting that let-7 is necessary for neuronal maturation. Taken together, the data reveal that cells lacking let-7 do differentiate into interneurons, however, they fail to both radially migrate into the OB layers and to fully mature [40].
Since a previous study had shown a role for let-7 in neuronal autophagy [66] we investigated if autophagy-related genes are controlled by let-7 in newborn OB-neurons by conducting Argonaute2-RNA Immunoprecipitation (AGO2-RIP) followed by qRT-PCR. We found that two autophagy-related genes, Slc7a5 and Slc3a2, are direct let-7 targets in newborn OB interneurons [40]. These findings suggest that loss of let-7 in newborn neurons could lead to altered neuronal autophagy, which would thereby impact radial migration. To investigate autophagy levels in newborn neurons lacking let-7, we used electron microscopy on let-7-knock-down cells and identified that there were fewer and smaller autophagic structures in cells lacking let-7 compared to control cells, suggesting decreased levels of autophagy. Immunohistochemical staining for sequestosome-1 (p62) showed increased levels associated with the lack of let-7. p62 is a cargo transport protein that is selectively degraded by autophagy, leading to its accumulation upon autophagy impairment and its decrease upon autophagy activation [84, 85]. Rescuing autophagy by co-injection of LV.let-7.sp together with transcription factor EB (TFEB) or Beclin-1 (Becn1) [86, 87], led to increased radial migration of interneurons into the different OB layers, thereby confirming that regulation of autophagy, through let-7, is a key process in adult neurogenesis.
CONCLUSIONS AND FUTURE DIRECTIONS: CROSSTALK BETWEEN MICRORNAS AND AUTOPHAGY IN NEURODEGENERATION
OB interneurons born during adult neurogenesis have been shown to differ from interneurons that are generated during embryonic neurogenesis in several of their properties such as morphology, position in the OB layers, electrophysiological properties and intercellular connections [22, 27, 88]. The function of adult-born interneurons in the OB remains disputed, however, a role in odour processing, olfactory learning and memory has been suggested and they have been implicated in improving the plasticity of neuronal networks [27, 33, 89]. In contrast to most other mammals, adult neurogenesis does not occur in the OB of the human brain. Instead, adult neurogenesis has been demonstrated to occur in the striatum [6, 18]. Due to obvious challenges regarding the study of the adult human brain, the origin of new neurons in the striatum remains unclear, however due to proximity, the SVZ has been suggested as potential source [4]. In line with this, adult-born rodent OB-interneurons share some similarities with adult-born human striatal neurons [34].
When adult-born interneurons reach the OB, they radially migrate from the centre of the OB to its periphery, where they mature and integrate into the pre-existing neuronal circuitry [22]. Functional integration of adult-born interneurons is a slow process [27] and is therefore likely to be tightly controlled. We identified let-7 as the most abundant miRNA in adult newborn OB interneurons, and as an important regulator of radial migration and maturation [40]. Loss of let-7 in newborn interneurons led to impaired levels of autophagy and activation of autophagy upon loss of let-7 rescued radial migration but not neuronal maturation, and hence revealed autophagy-dependent and independent let-7 functions in newborn OB interneurons. In addition, Morgado et al have recently found that neuronal differentiation of NSCs is controlled by miR-34a. Regulation of miR-34a is partially dependent on autophagy which further broadens the importance of miRNA-controlled autophagy in adult neurogenesis [63]. Together, these results reveal a link between miRNA regulation, adult neurogenesis and autophagy.
These observations are interesting in relation to neurodegenerative disorders such as HD, where an impairment of autophagy has been observed [73, 90–92]. In HD this impairment is characterised by a late stage block of autophagy, resulting in dysfunctional cargo loading and empty lysosomes [73, 92, 93]. Intriguingly, wild-type huntingtin (wtHTT) itself has been implicated in the regulation of autophagy by promoting autophagosomal transport and by acting as scaffold for selective autophagy [73, 94, 95]. In addition, mounting evidence demonstrate that miRNAs play an important role in neurodegenerative diseases [96–98]. Conditional deletion of Dicer in specific neuronal populations, such as excitatory forebrain neurons or dopaminergic midbrain neurons in the adult mouse brain, results in neurodegeneration [99–101]. In line with this, altered expression levels of individual miRNAs have been reported in Alzheimer’s, Parkinson’s and Huntington’s disease [96, 102]. For example, several studies have analysed miRNA levels in different models of HD and revealed altered expression levels of e.g miR-9, miR-124 and miR-132 [103, 104]. Furthermore, wtHTT has been demonstrated to interact with AGO1 and AGO2 in p-bodies, suggesting a direct role for wtHTT in post-transcriptional regulation [105].
In the light of our data on let-7, further studies are therefore warranted to investigate if the impairment of adult neurogenesis in HD and other neurodegenerative disorders is caused by an impairment of autophagy, which is linked to alterations in the miRNA-network. This would further strengthen the rationale for the development of autophagy activating treatments, perhaps through modulation of miRNA-levels. To date, boosting autophagy through pharmacological or genetic manipulation has successfully reversed disease-associated phenotypes in transgenic mouse models of HD and some other neurodegenerative diseases. Boosting autophagy in this manner has been shown to be associated with a reduction of the protein aggregate burden [74, 106, 107]. These pre-clinical findings have led to the first clinical trial of rilmenidine in mild HD, as a means to upregulate autophagy [108, 109]. While these initial studies have shown that this approach is feasible and well tolerated, it is also evident that therapeutic approaches to activate autophagy need to be optimized and tailored for different neurodegenerative disorders. A clear understanding of exactly how alterations in autophagy contribute to cellular dysfunction and death in neurodegenerative disorders is currently lacking. Our let-7 data [40] provide further support for developing autophagy-activating therapeutic approaches for HD and other neurodegenerative disorders, since they suggest that activation of autophagy will not only clear toxic protein aggregates, but also directly restore dysfunctional adult neurogenesis.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
AUTHORS CONTRIBUTIONS
K.P., R.P. and J.J. wrote and edited the manuscript. All authors read and approved the final manuscript.
Footnotes
ACKNOWLEDGMENTS
We are grateful to Deirdre Hoban for excellent comments on the manuscript and to Bengt Mattsson for valuable help with illustrations. The work in our laboratory is supported by grants from the Swedish Research Council, the Swedish Foundation for Strategic Research, the Swedish Brain Foundation; the Swedish excellence project Basal Ganglia Disorders Linnaeus Consortium (Bagadilico), and the Swedish Government Initiative for Strategic Research Areas (MultiPark & StemTherapy).