
Abstract
BACKGROUND:
Advanced urothelial carcinoma (UC) is an aggressive disease whose mutagenic processes are yet to be elucidated. Targeted therapies are urgently needed, but the road from bench to bedside is slowly progressing. In this review, we discuss urothelial carcinoma etiology, along with the most recent advances in UC candidate targeted therapies.
METHODOLOGY:
A comprehensive database search was performed. We aimed to review the most recent updates on UC genomics and targeted therapies. Pre-clinical as well as clinical studies were included.
RESULTS:
Our review highlights the advances in understanding the molecular basis of urothelial tumorigenesis, including smoking, chemical parasitic carcinogens, inheritance, and APOBEC3 editing enzymes. We discussed how these factors contributed to the current mutational landscape of UC. Therapeutic options for UC are still very limited. However, several promising therapeutic approaches are in development to leverage our knowledge of molecular targets, such as targeting fibroblast growth factor receptors (FGFR), DNA damage repair pathways, and HER2.
CONCLUSIONS:
Blindly testing targeted therapies based on other cancer data is not sufficient. UC-specific biomarkers are needed to precisely use the appropriate drug for the appropriate population. More efforts to understand UC biology and evolution are urgently needed.
INTRODUCTION
Urothelial carcinoma (UC) of the bladder affects 80,000 patients and causes 17,000 death annually [1]. Urothelial carcinoma of the bladder (UCB) is initially a chemo-sensitive malignancy with patients experiencing objective response rates of 40-60% with standard first-line cisplatin-based chemotherapy. However, response durations are typically short-lived with the median survival of patients with metastatic bladder cancer being ∼15 months. Phase I-III clinical trial studies have demonstrated that durable responses are achieved in 20-25% of patients with platinum-resistant metastatic UCBs treated with PD-1/PD-L1 immune checkpoint blockade (ICB) [2, 3]. Thus, while ICB has dramatically changed the landscape of treatment for metastatic UCB, only a minor portion of patients benefit from these treatments. Moreover, in the majority of cases, acquired resistance to ICB is inevitable, highlighting the need for predictive markers for response and patient-specific therapeutic strategies. The advances in molecular characterization of urothelial cancer revealed enrichment with actionable molecular targets such as Fibroblast Growth Factor Receptor 3 (FGFR3).
In this review, we will cover the advances in UC genomics and what has been achieved in answering the following key questions 1) what causes DNA genomic alterations in UC, 2) what was achieved to molecularly characterize the UC, and 3) interpretation of the driver and actionable mutations. To answer these questions we conducted a comprehensive database search till December 2022 including clinical and preclinical studies.
WHAT CAUSES DNA GENOMIC ALTERATIONS IN UC?
Several environmental factors were shown in epidemiological studies to contribute to the risk of bladder cancer [4, 5]. One of the innovative approaches investigating the molecular basis of carcinogens is to study mutational signatures. Mutational signatures are unique combinations of mutation types generated by distinct mutational processes. For example, tobacco smoking has a preferential pattern in inducing characteristic single-base substitutions (SBS). The preferential pattern of the mutagenic effect of smoking can be identified computationally from sequencing data. If a given tumor is enriched with a characteristic pattern of SBS, then a smoking-related mutational signature can be deduced. The Catalogue of Somatic Mutations in Cancer (COSMIC) initiative from the Sanger Institute provided a continuously updated source of mutational signatures in cancer. Not all of these signatures were linked to a definite carcinogen or causative mechanism.
Smoking is among the most significant risk factors with a relative risk of 3.5 [6]. The paucity of studies interrogated the molecular basis of these risk factors and the cascade of genomic alteration events that lead to urothelial cancer. One analysis of The Cancer Genome Atlas- bladder cancer (TCGA-BLCA) cohort showed that smokers have upregulation of G-protein coupled receptor 15 (GPR15) expression and enriched with mutations in SPTA1, TP53, KDM6A, and KMT2D compared to nonsmokers [7].
Aristolochic acid (AA), a mutagen and a compound of plant origin was linked to bladder cancer. AA-induced nephrotoxicity (AAN) has been extensively studied, with a recent correlation with earlier urothelial tumorigenesis and incidence of multifocal disease [8]. Typically, the sequalae are as follows; long-term exposure leads to AAN ending in chronic kidney disease, with development of urothelial carcinoma a few years following kidney transplantation. 14% of kidney transplantation patients with a history of AAN develop early multifocal urothelial tumors compared to 1-2% AA naïve transplantation patients [9, 10] suggesting an active effect of AA rather than the impact of transplant-associated immunosuppression. AA correlates with unique mutational signatures in the form of A:T<T:A transversions at CAG trinucleotide 3‘ splice sites [11, 12]. AA genotoxicity varies with ethnic, geographic, gender, and affected genes [13, 14], especially observed in TP53 [15, 16], HRAS [17], and KRAS [18]. Bellamri et al. [19] demonstrated dose-dependent gene damage in RT4 human bladder cell line exposed to AA, through varying pathways.
Not only chemicals are capable of causing bladder cancer, urinary tract infections (UTIs), and indwelling urinary catheters role in UC tumorigenesis is of question [20–22]. Other works suggested that urine is unsterile, concluding that urinary microbiota is a strong contributing factor [23, 24].
Schistosoma haematobium, a tropical chronic parasitic infection, has been closely linked to bladder cancer epidemics [25]. Though Schistosoma-induced BCa (SIBC) and non-Schistosoma-induced BCa (NSIBCa) differ in gene structure and histopathology [26, 27]. Both SIBC and NSIBC are managed according to the same guidelines [28]. The mechanisms by which Schistosoma haematobium results in bladder cancer are yet to be known. Some suggested that bacterial interactions with schistosomes promoted a state of chronic irritation and later on led to squamous metaplasia of the bladder urothelium [29]. Others suggested that soluble antigens of Schistosoma itself are the direct cause of genotoxicity, aided by chronic inflammation [30]. SIBC incidence is in decline owing to schistosomiasis infection control [31].
APOBEC3 cytidine deaminases have been implicated as the source of widespread mutagenesis in human cancers [32]. By mutating single-stranded DNA or RNA in the form of SBS mutations in clusters known as Kataegis [33], they induce neoantigens in all cancer types, with UC at the top of the list [34]. Cytosine replacement with thymine C:T; C:G<T:A signature, which predominates in bladder cancer and correlates with advanced UC stages, is caused by the APOBEC3 editing system [35–37]. These signatures are absent in urothelial papilloma and inverted urothelial papilloma of the bladder, which follow a benign disease course [38]. APOBEC3 correlates with high tumor mutational load [34]. It reshapes a given tumor genetics in a stochastic manner, contributing to neo-antigens formation [39]. With their inability to impact double-stranded DNA, they require assistance to affect the human genome. For example, APOBEC3B synergizes with hnRNP A3 (heterogeneous nuclear ribonucleoprotein A3) that recruits telomerase activity, while APOBEC3B induces mutations [40]. Supported by finding most mutated regions near telomers and centromeres [35] APOBEC3 signatures manifest in relation to the history of aristolochic acid (AA) exposure as well [41].
APOBEC3 enzymes are well known by their antiviral activity [42], which suggests UC induction by a viral agent through APOBEC3 activity, as observed in Human papilloma virus (HPV)-driven cervical carcinoma [43]. BK Polyomavirus (BKPyV), an infection of childhood, is accused of such a mechanism in the UC setting [44].
Some cancers and cancer syndromes are caused by inheritance [45]. Urothelial carcinoma heritability is still being studied, though it is already linked to heritable cancer syndromes as Lynch syndrome [46]. Inheritance happens due to processes that take place very early in life [47]. The Spanish Bladder Cancer Study showed that patients with newly diagnosed bladder cancer were more likely to be associated with a family history of cancer in≥1 relative (OR, 1.32; 95% CI, 1.11-1.59) [48]. Another Italian case-control study on bladder cancer showed that the OR for a family history of bladder cancer was 2.13 (95% C is: 1.02–4.49) [49]. A twin study that followed up 80,309 monozygotic and 123,382 same-sex dizygotic twin individuals for 32 years estimated a bladder cancer heritability of 30%, which was proximate to breast cancer [50]. Heritability in this study was defined as the proportion of variance in cancer risk due to interindividual genetic differences [50].
Two key studies characterized the pathogenic germline variants in UC patients using targeted sequencing of known cancer susceptibility genes. Carlo et al. used a pipeline “PathoMan” that prioritizes germline variants in 77 cancer predisposition genes from targeted sequencing data using MSK-IMPACT platform [51]. They identified 86 pathogenic or likely pathogenic variants affecting 80/586 (14%) of UC patients. The frequency of patients with pathogenic variants in DNA damage repair (DDR) genes was 11.3%. One-third of the pathogenic variants identified in DDR genes underwent loss of heterozygosity (LOH). Nassar et al. analyzed targeted clinical germline testing data of 1038 patients with high-risk UC covering a median of 42 genes [52]. The frequency of patients with pathogenic germline variants was found to be 24%. Patients with germline variants in DDR genes were 20%.
Some medications are an independent mutagenic factor. For example, anticancer chemotherapy contributes to cancer evolution. Cancer cells, heterogeneous and unstable, can easily reshape themselves to evade death. Some mutations are private to the primary tumor. Their absence from its metastases suggests their elimination by therapy, being “unfit” to combat its toxicity. This is called “selective pressure” [39]. However, the process of selective pressure is difficult to study. Currently, genomic research is unable to grasp the dynamics of genetic evolution. Whether the resisting clones are present from the start of therapy or develop against the given agent is still unclear. Field cancerization hypothesis and radical molecular subtype shifts, either can be a plausible explanation to this phenomenon. The multifocal nature of non-muscle invasive bladder cancer (NMIBC) generated field cancerization while gaining an aggressive behavior in a previously relatively benign condition suggested the gain of more/different driver mutations. Surprisingly, a fraction of patients has progressive disease while still gnomically stable. Predicting the prognosis of an NMIBC will better direct clinical efforts to avoid recurrence and progression [53].
MOLECULAR CHARACTERIZATION OF UROTHELIAL CANCER
Molecular characterization of urothelial cancer showed remarkable advancement in the last few years, starting with the comprehensive molecular analysis of the full TCGA cohort of 412 muscle-invasive bladder cancer (MIBC) released in 2017 [54]. Several high-throughput sequencing studies of urothelial cancer have been accomplished since then. We have compiled a dataset of individual patient genomic data from 2133 patients with urothelial cancer extracted from 14 studies using cBioportal. We identified 19 cancer-related genes with mutation frequency > 10%. The frequencies of top mutated genes are outlined in Fig. 1. Most of these samples are primary tumor tissue. The key cellular and molecular altered pathways were cell cycle regulations (TP53 and RB1), chromatin remodeling (KMT2D, KDM6A, ARID1A, KMT2 C, EP300, and KMT2A), receptor tyrosine kinase activity (FGFR3, PIK3CA, ERBB3, ERBB2), transcriptional coactivator (CREBBP and ELF3), chromosomal segregation (STAG2), and telomerase activity (TERT).
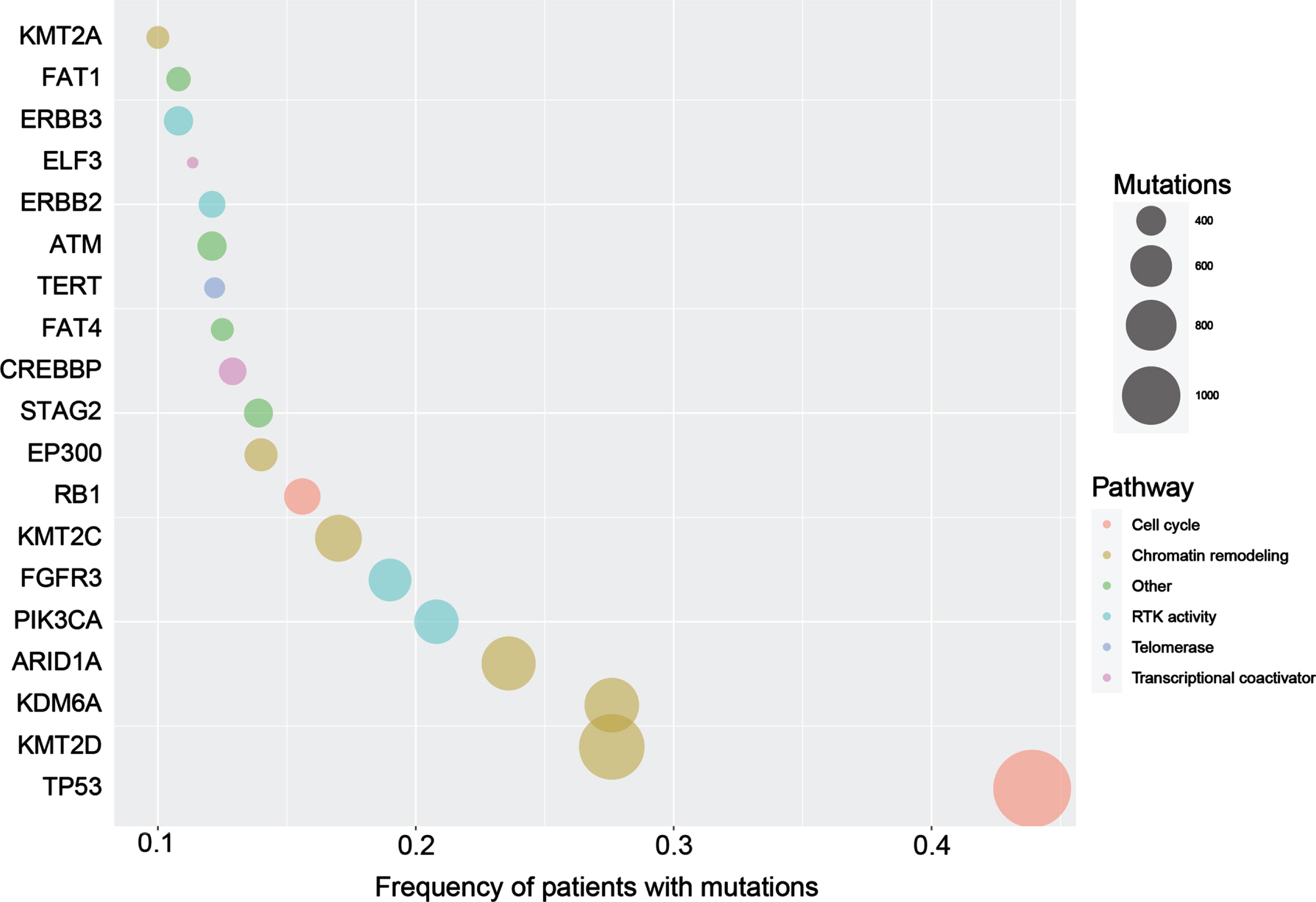
Bubble plot showing the genes with most frequent mutations in patients with urothelial cancer. The size of circles indicates the number of different mutations per gene.
Results of PAPR inhibitors in bladder cancer
*HRD: defined as genomic loss of heterozygosity (LOH)≥10%. platinum based chemotherapy (PBC).
Key studies investigating efficacy of targeting HER2 in bladder cancer
Two caveats hinder leveraging the large-scale genomic data of patients with UC. First, most of the altered genes occurred in less than 15% of patients (Fig. 1). Second, analyzing the total number of mutations affecting these genes showed that most of the mutations are singletons (Fig. 1). In other words, there is a paucity of recurrent mutations that allow developing customized hotspot genetic testing. In addition, the large variety of non-recurrent mutations makes it difficult to provide accurate functional annotations.
THE SPECIAL CASE OF BLADDER CANCER VARIANTS
There is limited data on the molecular profiles of bladder cancer variants. A study by Roy et al. showed less frequent aberrations in the chromatin-modifying genes and TERT promoter regions compared to conventional UC [55]. Another study, limited to the urachal subtype, confirmed the lower frequency of TERT promoter mutations (n = 1/23) [56]. The overall mutational profile of adenocarcinoma of the bladder is similar to colorectal adenocarcinoma with frequent mutations in KRAS, TP53, and SMAD4. In small case series, actionable targets such as EFGR, ERBB2, and BRAF were described [57, 58]. A case report described an objective response to an anti-EGFR agent (cetuximab) in a patient with EGFR amplified urachal adenocarcinoma [58]. An immunohistochemical study of 36 primary adenocarcinomas from cystectomy samples showed nuclear p16 and p53 expression occurring in 67% and 58% of the cases, respectively [59]. Whole-exome sequencing of 7 urachal adenocarcinomas showed recurrent NF1 mutations in three cases suggesting role for the MAP- kinase pathway [60]. The mutational analysis of 117 samples of squamous cell carcinoma of UB in the COSMIC database showed the mutation rate of CDKN2A and FGFR3 were 56% (35/63) and 10% (4/40, all p.S249 C), respectively [61]. Another study identified FGFR3 mutations in 8.5% (6/71, all p.S249 C) of squamous cell carcinomas of the bladder [62]. FGFR3-mutant tumors were associated with significantly shorter recurrence-free survival compared to non-mutant tumors (7.5 vs. 10.5 months) [62]. A study of 61 patients showed that squamous cell carcinoma of the bladder harbors a similar mutational profile compared to conventional urothelial carcinoma, including alterations in chromatin modifiers and TERT promoter regions [63]. Another study showed a high prevalence of APOBEC mutation signature, which is distinct from the tobacco-associated signature found in small cell lung cancer [63, 64].
HOW TO DEFINE THE DRIVER AND ACTIONABLE GENOMIC ALTERATIONS IN UC
Although UC is among cancers with high mutational load, identifying the cancer driver genes is challenging. The common approach for identifying cancer driver genes is through computational tools. The FGFR3 targeting therapy, Erdafitinib, is the only approved targeting agent in UC [65].
Neurotrophic tropomyosin receptor kinase (NTRK) encodes protein kinases, that are activated by gene alterations driving tumorigenesis [66, 67], most commonly, gene fusions, which were absent in UC on Pan-cancer analysis using TCGA [68], while 0.34% of bladder tumors harbored NTRK fusions using FoundationCore database [69]. NRTK-3 alterations suggest advanced UC and poor prognosis [70] NTRK alterations are rare, with fair response to TRK inhibitors regardless of tumor histology and patient population [71, 72]. They are under investigation in UC setting in combination with nivolumab PDL-1 in study NCT03606174 [73].
Despite the prevalence of HER-2 expression alterations in many cancer types, only breast and gastric cancers benefit from targeting it, suggesting that more than altered expression is needed to settle for HER-2 targeting therapy [74–78]. Although studies correlating HER-2 expression and BUC behavior show conflicting results [79, 80], Her-2 overexpression is a prognostic biomarker for recurrence and patient survival in NMIBC [81–84], but has not been a successful therapeutic target yet [85, 86]. One case study had a complete response to trastuzumab+gemcitabine as a third-line treatment [87], while others demonstrated the possibility of adding trastuzumab to chemotherapy with acceptable toxicity [88, 89]. Further preclinical and clinical studies are needed to cement the role of HER-2 in UC setting [90]. We emphasize the importance of standardizing the methodology by which alterations are detected to help case recruitment in clinical trials.
DDR truncating alterations are present in up to 30% of high grade NMIBC in one case series [91]. Therefore, it was worthwhile to test PARP inhibitors in patients with advanced UC. The ongoing trials evaluating PARP inhibitors either alone or combined with other agents are of two categories. First, the all-comers trials include a trial that investigates niraparib plus atezolizumab after the failure of platinum-based chemotherapy (NCT03869190) and another trial investigating Olaparib plus durvalumab (NCT03534492). Second, somatic-based biomarker trials, including a trial investigating durvalumab alone compared to durvalumab plus Olaparib in mutant HRR-selected patients (BAYOU trial). In addition, two trials investigating Olaparib monotherapy in patients with somatic alterations in DDR gene panels (NCT03448718 and NCT03375307). Unfortunately, PARP inhibitors failed to show significant clinical activity in advanced UC either alone (ATLAS trial) [92] or with immune checkpoint blockade (ICB) (BISCAY trial) [93].
A major challenge of these studies is using a highly variable set of DDR genes or non-validated phenotypic biomarkers, preventing robust conclusions. The prioritized variants and genes are extrapolated from data of other cancers, which are not necessarily specific to UC. Ongoing trials lacked the distinction between germline and somatic variants in DDR genes. Somatic and germline DDR mutations are not created equal. The assumption that somatic DDR gene alterations are a tumor-agnostic biomarker for DNA damaging agents is not supported by evidence. In fact, a recent analysis of BRCA1/2 mutations showed that phenotypic consequences, as assessed by somatic LOH and HRD signature, are tumor-lineage specific and thus are potentially different in UC compared to other cancers [94]. The ideal genetic biomarker has to identify patients with variants that were proven to have clear functional and clinical consequences. Likely, the lack of patient selection based on germline testing of DDR mutations in these trials led to these negative results. Still, it is more likely to demonstrate efficacy signal in somatic biomarker-based trials (e.g., BAYOU trial) compared to all comers trials.
Vosoughi et al. propose that germline alterations in DDR as defined by robust computational frameworks would be a reliable biomarker to select patients with UC to PARP inhibitor. In addition, the selection of patients with prior platinum sensitivity or platinum-naïve is worth prioritizing. They have developed a computational framework to identify putative deleterious germline variants (pDGVs) from whole-exome sequencing (WES) data of germline DNA and 157 primary and metastatic tumors from 80 UC patients [95]. They identified pDGVs in DDR genes in 7.5% of patients with advanced UC. Furthermore, it has been shown that germline variants undergo LOH at a high rate and play a critical role in metastatic progression through progressive somatic LOH and extensive germline-somatic interactions [51, 95].
Interestingly, exceptional responders to PARP inhibition are more frequently harboring germline alterations. For instance, one patient in the ATLAS trial was evaluated and found to have BRCA1 truncating germline mutation. The patient achieved stable disease with approximately 10% tumor reduction on Olaparib. Other case reports in the literature reported patients with germline alterations achieving durable responses [96, 97].
Apart from using the right biomarker, there is a need to characterize the PARP inhibitors resistance mechanism in UC, including DDR rewiring and PARP1 mutations. In addition, germline-somatic genomic interactions generate additional therapeutic vulnerabilities identifying drug combinations that synergize with PARP inhibitors. These are critical endeavors needed to improve outcomes for patients with metastatic UC harboring DDR mutations.
CONCLUSION AND FUTURE DIRECTIONS
In summary, there are multiple candidate targets for drug development to be considered for urothelial cancer patients. New approaches are needed to help select patients in given trials. Next generation sequencing (NGS) is a realistic solution when it comes to choosing targeted therapies, on the bench and at the bedside. However, moving beyond tumoural DNA alterations is crucial specifically leveraging transcriptomics, metabolomics, and microbiome as drug targets. Efforts toward validating biomarkers for UC are needed to obtain better results. A better understanding of mutagenesis promises better-targeted therapies in the near future. For example, early branching theories and the diversity of tumor clones suggest that targeted therapies are beneficial early on. This will be better tested through developing new techniques to modulate mutagenesis such as APOBEC enzymes or specific intratumoral or intracellular microbiota.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments.
FUNDING
The authors report no funding.
AUTHOR CONTRIBUTIONS
RMA conducted the review of the literature and wrote the first draft of the manuscript. All authors contributed to the conception and interpretation of data. All authors approved the final version of the manuscript.
CONFLICT OF INTEREST
KSS is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review. RMA, EY and AHR have declared no conflicts of interest.