
Abstract
BACKGROUND:
For decades, immunotherapies have been integral for the treatment and management of bladder cancer, with immune checkpoint inhibitors (ICIs) transforming patient care in recent years. However, response rates are poor to T cell-targeted ICIs such as programmed cell death protein 1 (PD-1) and programmed cell death-ligand 1 (PD-L1) blocking antibodies, framing a critical need for complementary immunotherapies. Promising strategies involve harnessing the activation potential of natural killer (NK) cells. They quickly exert their antitumor activity via signaling through germline-encoded activating receptors and are rapidly sensitized to new tissue microenvironments via their regulation by polymorphic HLA class I, KIR and NKG2A receptors.
OBJECTIVE:
In this review, we examined the roles of currently available NK-targeted antitumor treatment strategies such as engineered viral vectors, small-molecule IMiDs, NK agonist antibodies, interleukins, and chimeric antigen receptor (CAR) NK cells, and their potential for improving the efficacy of immunotherapy in the treatment of bladder cancer.
METHODS:
Through review of current literature, we summarized our knowledge of NK cells in solid tumors and hematologic malignancies as their roles pertain to novel immunotherapies already being applied to the treatment of bladder cancer or that offer rationale for considering as potential novel immunotherapeutic strategies.
RESULTS:
NK cells play a critical role in shaping the tumor microenvironment (TME) that can be exploited to improve T cell-targeted immunotherapies.
CONCLUSIONS:
Emerging evidence suggests that NK cells are a prime target for improving antitumor functions in immunotherapies for the treatment of bladder cancer. Further research into profiling NK cells in settings of immunotherapies for bladder cancer could help identify patients who might maximally benefit from NK cell-targeted immunotherapies and the various approaches for exploiting their antitumor properties.
INTRODUCTION
The first instance of immunotherapy in bladder cancer was observed in 1976 when Morales et al. demonstrated the effectiveness of intravesical Bacillus Calmette-Guerin (BCG) instillation in the treatment of superficial bladder tumors [1]. Since then, significant strides have been made in the advancement of bladder cancer immunotherapy, in the realm of immune checkpoint inhibitors (CPI). The well-known PD-1:PD-L1 axis is a popular target in the treatment of both non-muscle invasive (NMIBC) and muscle-invasive bladder cancer (MIBC), and several monoclonal antibodies have been approved for this purpose. However, the overall response rates to these T cell-targeted CPI remain low, and additional strategies must be considered [2–5].
Alongside T cells, natural killer (NK) cells are also critical effector cells in an antitumor immune response and among the earliest responding cells following tumorigenesis. Tumor surveillance by NK cells is achieved through a program of education, whereby interactions between NK cell receptors and HLA class I ligands sensitize NK cells to distinguish between diseased and healthy cells and recognizing perturbed expression of HLA class I [6, 7]. This process is regulated by an immunogenetics system that combines conserved CD94:NKG2A and NKG2C receptors that recognize non-polymorphic HLA-E and the variable killer cell immunoglobulin-like receptors (KIR) that recognize polymorphic HLA-A,-B and -C [8]. These inhibitory interactions are balanced by the combinatorial expression of activating receptors on NK cells resulting in NK cell phenotypic diversity and functional sensitivity that vary considerably within an individual, and have major functional consequences on host immunity to infection and cancer [9–11]. Emerging evidence indicates that loss of HLA class I expression on tumor cells is a common phenotype associated with many cancers [12–17], including bladder cancer [18] (Fig. 1), and suggest that HLA class I genotype may predict tumor response to immune checkpoint blockade (ICB) targeting PD-1 or PD-L1 [19–21]. The human immune system relies on HLA class I to present antigens to CD8 T cells while modulating NK cell education and functional sensitivity.
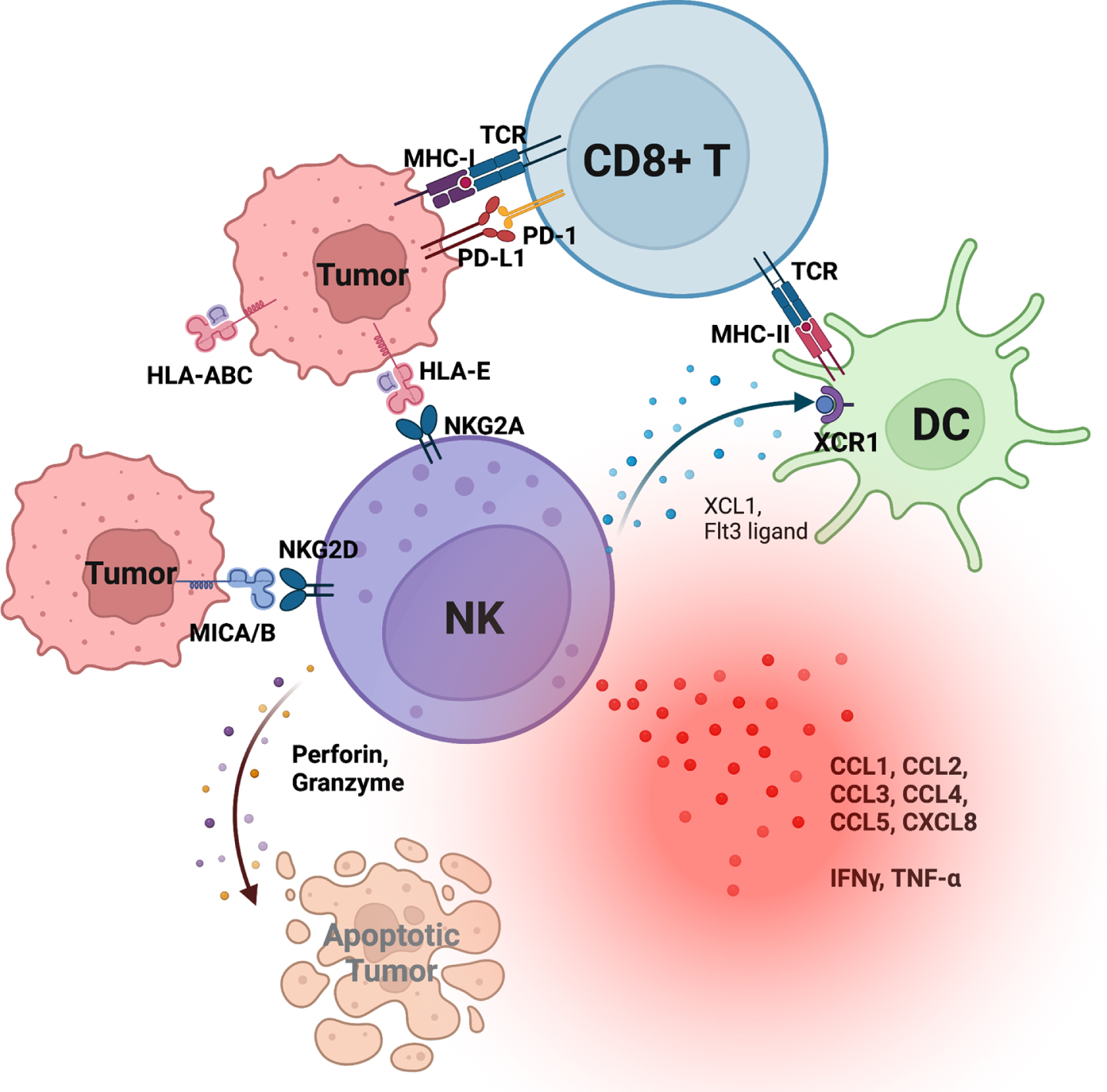
Schematic representation of tumor-infiltrating natural killer (NK) cells and their interactions with tumors and dendritic cells (DC). NK cells provide critical cytokines and chemokines that recruit and activate DCs, which subsequently recruit and activate CD8 T cells.
NK cells are members of the lymphoid lineage, which additionally includes γδ T cells, invariant NKT cells, regulatory T cells, CD4 and CD8 T cells, and B cells [22]. While NK and CD8 T cells express an overlapping array of functional receptors, a canonical CD8 T cell response is defined by exquisite specificity through T cell receptors (TCR) engaging a peptide/HLA class I complex as well as required co-stimulatory signals through CD28 and cytokine receptors [23]. Conversely, NK cells can generate a rapid response by forming a net-effective signal from activation of their germline-encoded receptors. This property allows NK cells to react as early, sentinel cells with helper and cytolytic antitumor properties. Though NK cells make up only a small fraction of the total blood lymphocytes, they may reside in larger numbers in lymphoid and peripheral tissues, making them an attractive target to exploit for immunotherapeutic potential [24, 25].
NK cells shape adaptive immunity through crosstalk within their tissue microenvironments
Early on following tumorigenesis, NK cell infiltration into the tumor microenvironment (TME) is critical for subsequent CD8 T cell recruitment and activity (Fig. 1). This sequence is observed in CPI-responsive mouse melanoma, where the efficient infiltration of CD8 T cells is dependent on prior NK cell infiltration [26, 27]. NK cell production of critical cytokines Flt3-ligand and XCL1 facilitates crosstalk with antigen cross-presenting XCR1+ dendritic cells (DCs) to enhance recruitment of CD8 T cells with antitumor functions. Upon engagement of their activating receptors with tumors, NK cells not only release cytotoxic substances for direct cytolytic killing, but they also secrete chemokines and cytokines that shape the TME for downstream activation of T cells and DCs [28]. Beyond provision of XCL1 and Flt3-ligand, NK cell-derived cytokines and chemokines include, but are not limited to, interferon-γ (IFN-γ), tumor necrosis factor-α (TNF-α), granulocyte macrophage colony-stimulating factor (GM-CSF), and stimulatory chemokines (CCL1, CCL2, CCL3, CCL4, CCL5, and CXCL8) [28]. Collectively, there is a large body of evidence indicating a critical ‘helper’ role in shaping adaptive antitumor immunity.
Interferon (IFN)-γ and tumor necrosis factor (TNF)-α are two hallmark effector cytokines that are critical in directing activation of immune cells. The IFN-γ receptor (IFNGR) consists of two subunits with distinct functions. IFNGR1 (α-subunit) is constitutively expressed on all nucleated cells and is involved in ligand binding, while IFNGR2 (β-subunit) expression is inducible and participates in downstream activation of Janus kinases (JAKs) and signal transducers and activators of transcription (STATs). Activation of IFNGR requires both IFNGR1 and IFNGR2, and triggers both antitumor and pro-tumorigenic processes in the TME [29]. In addition to inducing major histocompatibility complex class I (MHC-I) and MHC-II expression on antigen presenting cells (APCs) as well as on tumors, IFN-γ also promotes recruitment of phagocytic cells [30]. However, this antitumor cascade may be met with concurrent inhibitory signaling, as IFN-γ also upregulates PD-1 and PD-L1 expression on CTLs, expansion of regulatory T cells (Tregs), and Qa-1 on murine tumors and HLA-E expression on human tumors [31–34]. Our group recently demonstrated that HLA-ABC and HLA-E expression by bladder tumors decreases with cancer progression, highlighting the importance of TCR-independent antitumor processes as well as reliance of CD8 T cells on crosstalk with other immune cells, including but not limited to NK cells and DCs. [18]
Strikingly, NKG2A expression by CD8 T cells is induced after acquisition of PD-1 and most other immune checkpoints and reflects a temporal and functional divergence from classical exhaustion pathways, whereby NKG2A+ CD8 T cells appropriate activating receptors traditionally ascribed to NK cells allowing them to react to HLA class I-deficient tumor cells in a TCR-independent manner. Among these receptors, the DNAX-activating molecule-1 (DNAM-1) offers an important activating signal as demonstrated by DNAM-1-blocking abrogating tumor reactivity by NKG2A+ CD8 T cells. Importantly, the abundance of these cells in bladder tumors of patients with MIBC and metastatic urothelial carcinoma predicts prognosis and survivorship and further associates with responsiveness to PD-L1 blockade [18]. Where somatic mutations in tumor cells driving LOH is a common phenotype of many tumors, most available data are poorly curated with respect to stage of disease and treatments previously received. Additionally, class I HLA cell-surface (and mRNA) expression by bladder tumors was significantly diminished in all the screened MIBC patients, suggesting that somatic mutations, alone, driving LOH are not sufficient to explain this observation and other post-translational modifications affecting expression are likely at play [18].
Immunotherapies: Beyond checkpoint inhibitors
Crosstalk between NK cells, DCs, and T cells in the TME, the role of NK cells in both direct antitumor cytotoxicity as well as their potential in augmenting current and future immunotherapies via stimulation of DCs and T cells is only starting to gain appreciation. Thus, strategies to explore NK-based cancer immunotherapies are of great importance [28]. In this review, we will explore immunotherapies currently in the preclinical and clinical stages designed to enhance recruitment, activation, and strength of NK cell activity that can act as either standalone therapies or as complements to current available CPIs in the treatment of bladder cancer.
Viral vectors
Engineered viral vectors are promising tools in bladder cancer therapy and have the advantage of delivering their effects in a localized manner directly within the bladder. Two adenovirus-based therapies have undergone clinical trials. CG0070 is an oncolytic, tumor-selective adenovirus encoding a transgene expressing human GM-CSF, a cytokine that promotes activation and maturation of myeloid cells and is critical for DC development, differentiation, and survival [35, 36]. The tumor-specificity of CG0070 lies in its E2F-1 promoter, which controls selective E1A (E1a) gene expression primarily in cells with defects in the retinoblastoma (Rb) pathway as seen in several cancers [36]. E3-dependent production of GM-CSF is reliant upon E1a expression and results in tumor-selective expression of GM-CSF [36]. A phase I in-human study of CG0070 demonstrated a favorable safety profile, with bladder toxicity grades ≤2 as the most common adverse events (Table 1) [37]. Treatment with CG0070 resulted in high levels of GM-CSF in patient urine and led to complete response rates of 47% to 50% with 6 to 10.4 months median duration in patients that failed BCG therapy 12 to 24 months prior to initiation of CG0070 [37, 38]. Currently, two ongoing clinical trials are investigating the efficacy of CG0070 in combination with pembrolizumab (NCT04387461) or CG0070 alone (NCT04452591) in NMIBC patients.
Important trials in NK-based cancer immunotherapy
Adstiladrin (also referred to as instiladrin/Nadofaragene Firadenovec) is another replication-deficient, recombinant adenovirus-based therapy that encodes human interferon-α2b IFNA2B (IFNα2b), a mediator of the type I IFN response and a potent modulator of NK cell activation and subsequent antitumor activity [39, 40]. Although the bladder urothelium is typically lined by glycosaminoglycans (GAG) that hinder viral transduction, the inclusion of the polyamides Syn-3 in adstiladrin has greatly improved transduction efficiency, leading to detectable IFNα concentrations in patient urine [41–43]. Like CG0070, adstiladrin offers no dose-limiting toxicities and has a favorable safety profile according to phase I study results (NCT01162785) (Table 1) [43]. Further, data from Phase II studies of BCG-unresponsive patients showed 35% of patients achieved 12-month recurrence free survival (NCT01687244), and phase III data demonstrated 45.5% complete response maintained at 12 months (NCT02773849) [44, 45].
Where NK cells are long established as key mediators of antiviral immunity, there are virtually no data profiling immune microenvironmental changes in bladder tumors of patients treated with GC0070 nor adstiladrin. It will be critical for understanding mechanisms of secondary resistance and for improving patient selection criteria.
Small molecule NK modulators
Immunomodulatory drugs (IMiDs) have been shown to enhance NK cell activity [46]. IMiDs such as thalidomide derivates lenalidomide (CC-5013) and pomalidomide (CC-4047) enhance NK cell-mediated cytotoxicity by stimulating T cell-derived IL-2 and IFN-γ production, leading to increased numbers of circulating NK cells and promoting their activation [47]. Additionally, the presence of antitumor antibodies can create a synergistic effect with IMiDs to enhance antibody-dependent cellular cytotoxicity [47]. Lenalidomide has demonstrated efficacy in treating hematologic malignancies and is currently FDA approved as maintenance therapy for post-transplantation multiple myeloma patients and for the treatment of previously treated follicular lymphoma and marginal zone lymphoma in combination with rituximab [48]. Phase I studies of lenalidomide in solid tumors including bladder, renal, prostate, hepatocellular, pancreatic, melanoma, thyroid, and head and neck cancers either administered alone or with synergistic tumor-targeting antibodies have demonstrated variable tolerability and toxicity depending on dosing and combination therapy that was used, with the most common toxicities including cytopenias and myelosuppression [49–54]. The utility of lenalidomide in bladder cancer is not yet clear due to limited progression of clinical trials (NCT01373294, NCT01342172, NCT01352962), although a preclinical study showed that lenalidomide in combination with BCG can successfully reduce tumor burden in a mouse model of bladder cancer [55]. Like lenalidomide, pomalidomide has also demonstrated clinical response in patients with relapsed and refractory multiple myeloma. However, evidence for its efficacy is limited in solid tumors, and an early phase I study in patients with advanced solid tumors showed dose-dependent toxicities that included neutropenia, transaminitis, and gastrointestinal symptoms (NCT00482521) [56].
A second strategy for enhancing NK cell function is by inhibiting NK-suppressive pathways in the tumor microenvironment, one of which is the TGF-β pathway [46]. Vactosertib (TEW-7197) and galunisertib (LY21557299) are two small molecule inhibitors targeting the TGF- β type 1 receptor kinase, and both have been used in a number of phase I/II studies in the treatment of glioma, glioblastoma, pancreatic cancer, hepatocellular carcinoma, and other solid malignancies both as monotherapy and in combination with chemotherapy or CPIs [57]. Currently, a phase II, open-label, non-randomized study is underway that will examine response to vactosertib and durvalumab combination therapy in urothelial cancer patients who failed previous anti-PD-1 or anti-PD-L1 therapies (NCT04064190). It should be noted that TGF-β synergizes with a number of other cytokines leading to pleiotropic functions that may promote certain desirable functions. For instance, where TGF-β associates with more aggressive bladder cancer [58], it also induces NKG2A expression on PD-1+ CD8 T cells, in vitro, when in presence of IL-15. As recently demonstrated by our group, NKG2A+PD-1+ CD8 T cells were more proliferative and produced greater amounts of IFN-γ, TNF-α, XCL1, and IL-2 in response to class I HLA-deficient tumor lines [18]. Whether TGF-β and IL-15 co-exist temporally in bladder tumors has not been formally studied.
Agonist pathways
NK cells are characterized partly by their expression of several important activating receptors, including but not limited to the FcγRIIIA (CD16) receptor, which is critical for NK cell-driven antibody dependent cellular cytotoxicity (ADCC). Additionally, NK cells express natural group 2D (NKG2D) and natural cytotoxicity receptors (NCRs) NKp30, NKp44, and NKp46, as well as members of the signaling lymphocyte activation molecule (SLAM) family [59–61]. It is established that NK cells require a concerted engagement across these activating receptors to realize their full activation potential [62, 63]. NK cells also possess a number of co-stimulatory molecules [64]. For purposes of this review, we focus on 4-1BB (CD137), OX40, GITR, and CD27 as these molecules are already being targeted clinically for treatment of hematologic malignancies and advanced solid tumors. These molecules are part of the tumor necrosis factor receptor superfamily (TNFRSF) and are important for enhancing an antitumor response (Fig. 2) [46]. While CD27 is constitutively expressed on immature NK cells, naïve and memory differentiated T cells, and memory B cells and plasma cells, 4-1BB, OX40, and GITR are upregulated only after activation of effector cells [65, 66].
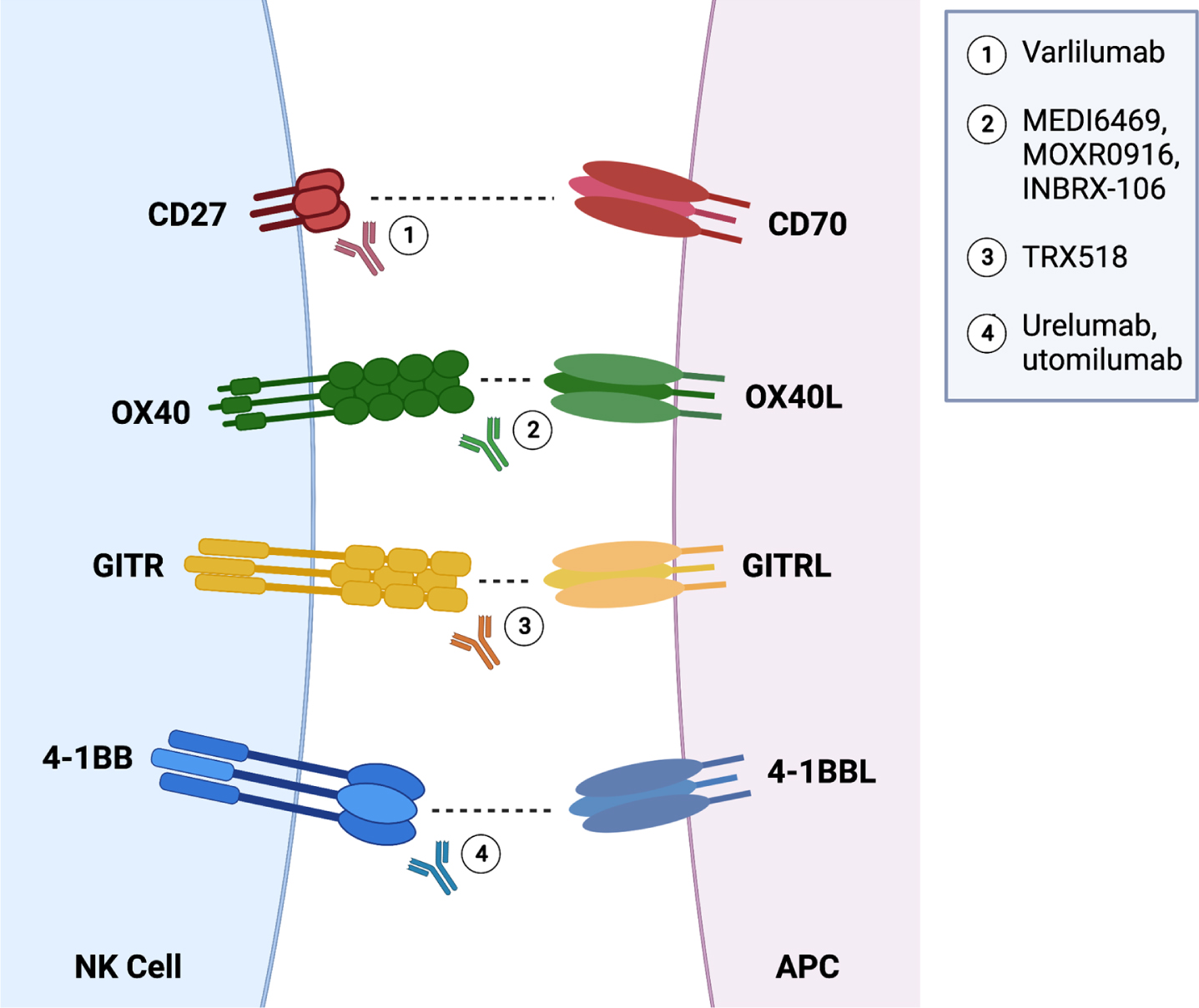
Schematic representation of activating (and potentially inhibitory) interactions between NK cells and antigen-presenting cells along with agonist antibodies being used for treatment across cancers.
The expression of 4-1BB on T cells is upregulated after TCR engagement, leading to upregulation of NF-κB signaling and pro-survival genes [67]. The efficacy of agonist antibodies against 4-1BB is highly dependent on CD4 and CD8 T cell activity, and these antibodies have demonstrated success in reducing tumor burden in mouse models of renal cell carcinoma, glioma, colon cancer, melanoma, and lymphoma [67]. The effects of 4-1BB ligand (L) on NK cells remains more elusive. It is established that culturing NK cells in presence of stimulator cell lines expressing 4-1BB-L increases gene expression of GZMB (granzyme B), PRF1 (perforin), and FASLG (Fas-L), yet this does not lead to improved killing of HLA class I-deficient K562 tumor lines [68]. In support of 4-1BB effects on NK cells, in vivo 4-1BB blockade in mice demonstrated a significant loss in NK cell numbers, possibly due to excessive IFN-γ release and 4-1BB–/– mice exhibit significantly reduced numbers of NK cells as well compared to their wildtype counterparts [69–72]. However, most recently, Li and colleagues demonstrated that signaling through the activating NKG2D receptor by NK cells in presence 4-1BB antibody and IL-27 resulted in enhanced cytotoxicity against prostate tumors [73]. This immunotherapy correlated with reduced expression of inhibitory KIR receptors, although phenotyping analyses were limited.
Phase I studies of the 4-1BB agonist urelumab (BMS-663513) in patients with B cell lymphoma revealed concerning hepatotoxicity both as monotherapy (NCT01471210) and in combination with rituximab (NCT01775631), especially at higher doses (Table 1) [74, 75]. Additionally, urelumab in combination with rituximab did not demonstrate improvements compared to rituximab alone [74]. On the other hand, urelumab in combination with cetuximab in head and neck cancer (NCT02110082) showed improved NK cell survival, DC maturation, and tumor antigen presentation, suggesting there may be value in investigating this further [76]. Utomilumab (PF-05082566) is another anti-41BB agonist antibody and has a better safety profile compared to urelumab due to its weaker agonist activity [77]. In a phase I trial when combined with rituximab (NCT01307267) in the treatment of relapsed or refractory follicular lymphoma and CD20+ non-Hodgkin lymphomas, no dose-limiting toxicities were observed and adverse events were limited to grades 1-2 [78, 79]. Utomilumab also showed a favorable safety profile when combined with pembrolizumab in the treatment of advanced solid cancers (NCT02179918) [80]. However, utomilumab may not be effective in patients who have previously failed CPIs [77].
OX40/CD134 acts as a co-stimulatory receptor on T cells, but its role on NK cells is much less understood. It was recently demonstrated that OX40 was induced on mouse and human NK cells in B-cell lymphoma, where OX40 expression was dependent on cell-to-cell contact between NK cells and monocytes and/or T cells [81]. However, where stimulation with anti-CD134 antibodies induced IFN-γ production and cytotoxicity by NK cells, CD134-ligand (CD134-L) did not. Further, combination therapy of anti-CD134 Abs with anti-CD20 Abs produced durable remissions, the response was dependent on CD16-signaling suggesting a critical synergy was required for effective antitumor immunity.
OX40 antibodies are also in clinical development. MEDI6469, a mouse-based OX40 antibody, has been studied in several tumor types, including B cell lymphomas (NCT02205333), prostate cancer (NCT01303705), breast cancer (NCT01862900), head and neck cancer (NCT02274155), and other solid malignancies (NCT01644968) (Table 1) [65]. A phase I in-human trial (NCT02274155) in locally advanced head and neck squamous cell carcinoma (HNSCC) showed that MEDI6469 was well tolerated and did not delay surgery [82]. MOXR0916, a humanized anti-OX40 agonist, has also demonstrated a favorable safety profile with no maximum tolerated dose and grade 1-2 adverse events (NCT02219724) [83]. A phase Ib trial of MOXR0916 with atezolizumab (NCT02410512) was recently completed and is currently undergoing phase I trials in solid tumors [65]. Finally, a phase I study investigating a new OX40 agonist antibody INBRX-106 in combination with pembrolizumab in solid tumors, including bladder cancer, is currently recruiting (NCT04198766) (Table 1).
The glucocorticoid-induced TNFR-related (GITR) protein plays an important role in immune surveillance and on the regulation of NK cell activation. Curiously, published data suggest that GITR may mediate different signaling pathways with opposing effects in mice and in humans, but ultimately impairs human NK cells, framing an important consideration for immunotherapy strategies targeting GITR signaling [84–87]. Not much is known about GITR expression by NK cells in solid tumors, but studies of hematologic malignancies, such as chronic lymphocytic leukemia (CLL) demonstrate that NK cells express higher levels of GITR in CLL patients compared with healthy controls [88]. GITR-GITRL interactions were studied in this setting and leukemia cells were shown to produce IL-6, IL-8, and TNF-α following GITRL-signaling post-culture with NK cells, leading to impaired degranulation, cytotoxicity, and production of IFN-γ by NK cells. Likely relevant to solid tumors, tissue resident/infiltrating monocytes produce these cytokines with established effects on promoting differentiation of NK cells, which includes down-regulating several important activating receptors, including but not limited to NKp46 and NKG2D [89, 90]. Thus, when considering immunotherapies targeting GITR, it will be important to profile any consequential negative effects on NK cells in the tumor microenvironment. Pre-clinical studies are actively engaged in overcoming the limited efficacy of GITR agonist antibody monotherapies. Most recently, when combined with anti-PD-1 as a bi-specific, mice (including a xenograft humanized mice) treated with anti-PD-1/GITR-L demonstrated growth inhibition of MC-38 colon carcinoma cells [91]. Interestingly, the mechanism of action was distinctly different compared with treatment of single agents alone or when in a 1:1 combination suggesting the bi-specific might directly target cells expressing GITR and high levels of PD-1, thereby excluding NK cells as targets.
In its first in-human phase I trial (NCT01239134), TRX518, an anti-GITR agonist antibody, has been reported to be safe without dose-limiting toxicities or treatment-emergent adverse events (Table 1) [92]. The latest reported phase I trial (NCT02628574) evaluated TRX518 in treatment-resistant, relapsed, or refractory metastatic cancers both as a monotherapy and in combination with other agents including gemcitabine, pembrolizumab, or nivolumab [93]. Although TRX518 maintained its previously reported safety profile, clinical response was limited in this patient population that received significant prior therapies [93].
In contrast to the aforementioned co-stimulatory molecules, the role of CD27 in antitumor activity is not as clearly defined, as the binding of CD27 to its ligand CD70 can result in both stimulatory and inhibitory effects. The stimulatory effects of the CD27-CD70 interaction are mediated by activation of NF-κ β and c-Jun pathways, leading to improved survival and proliferation of effector cells [65]. However, continuous expression of CD70 has been associated with increased Treg infiltration in tumors, consequently weakening the antitumor response [94, 95]. Varlilumab (CDX-1127), an anti-CD27 agonist antibody, has demonstrated favorable safety profiles in studies involving several solid and hematologic malignancies (NCT02284971, NCT02270372, NCT01460134) (Table 1) [96]. Varlilumab was well-tolerated when administered in combination with nivolumab, though its clinical utility may be limited as the overall response was not superior to that of nivolumab alone [97].
Important for understanding the effects on NK cells, CD27 is uniquely expressed by immature (CD56bright) human NK cells and to a lesser extent by transitioning intermediate mature NK subsets. These NK cells predominantly lack expression of CD16 as well as cytolytic machinery but instead are very sensitive to cytokines and chemokines. As NK cells mature, they acquire the capacity to produce granzymes, perforins and other mediators of cytolysis. And, the relative abundance of immature and mature NK cells vary significantly across tissues [25]. To the best of our knowledge, no one has considered the effects of promoting NK cell maturation and differentiation in immunotherapy strategies integrating anti-CD27 agonist antibodies.
Great efforts have been made to better understand mechanisms underlying secondary resistance to these immunotherapies. Design of combination immunotherapy strategies targeting NK cells offer an important and largely untapped effector cell either to target alone or rather as strategies for dually targeting NK and CD8 T cells.
Interleukins
NK cells are very sensitive to interleukins. Perhaps most prominently targeted for cancer immunotherapies is interleukin-15 (IL-15), which plays an integral role in NK and T cell development, activation, and proliferation [98]. Native IL-15 complexes with its α chain receptor IL-15Rα, which stabilizes the trans-oriented interaction between IL-15/15Rα and IL-2 receptor βγ chain (IL-2RβγC) on NK and T cells to allow for subsequent immune cell activation (Fig. 3, upper left plot) [98]. This interaction is enhanced by the IL-15 variant IL-15N72D, which contains an asparagine-to-aspartic acid mutation at amino acid 72 (N72D) [99]. ALT-803 (N-803) is an IL-15 superagonist made from the fusion of IL-15N72D and the extracellular IL-15Rα sushi domain (IL-15RαSu) to the Fc domain of IgG1 (Fig. 3, upper right plot). Compared to native IL-15, N-803 has the added benefits of a longer half-life and stronger binding activity to IL-2RβγC, both resulting in improved stimulation of NK cell activity and establishing an antitumor response [100, 101]. Combination therapy with N-803 and BCG demonstrated greater antitumor activity than either agent alone, and a phase Ib trial (NCT02138734) of the N-803/BCG combination showed no dose-limiting toxicities and only low grade (<3) adverse effects in BCG-naïve patients with NMIBC (Table 1) [102, 103]. Moreover, patients from the phase Ib trial (n = 9) remained disease free without progression 6 years after treatment, and a phase II/III trial (NCT03022825) for BCG-unresponsive patients is currently ongoing with promising interim results. Importantly, where BCG therapy induces strong activation and recruitment of NK cells and CD8 T cells to the bladder TME, patients repeatedly exposed to many doses of BCG experience sustained activation leading to immune exhaustion (Fig. 3, lower plot) [104, 105].
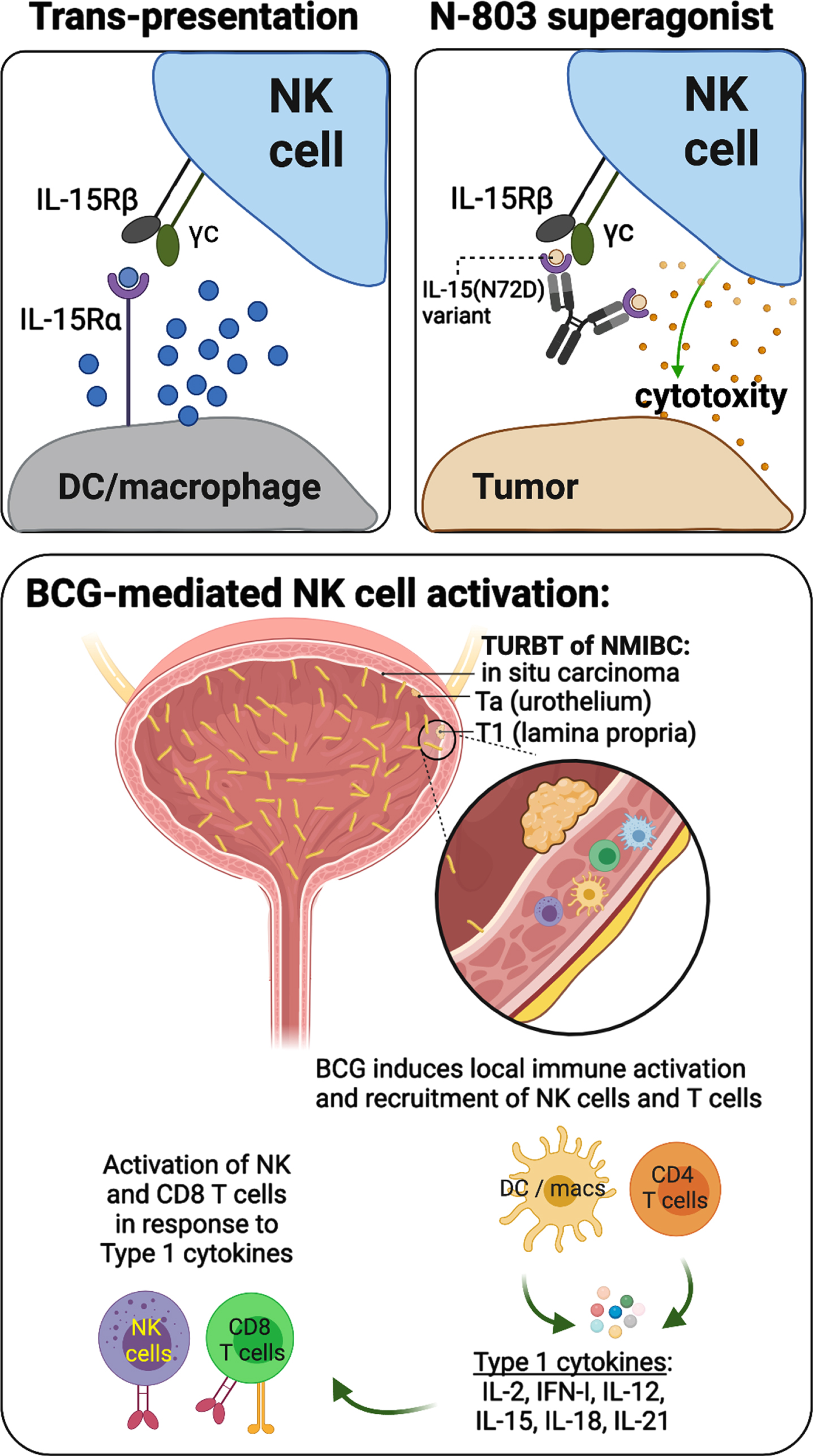
Schematic representations describing the mechanisms of IL-15 trans-presentation and activation of NK cells (upper left), the mechanisms of action underlying N-803 IL-15/IL-15Rα superagonist (upper right), and a working model of BCG-mediated activation of NK and CD8 T cells, which relies on indirect stimulation by dendritic cells (DCs), macrophages (macs), and CD4 T cells through production of type 1 cytokines.
In addition to IL-15, IL-7 has also demonstrated potential in antitumor therapy due to its function in increasing NK, CD4 T, and CD8 T cell proliferation in settings of metastatic breast cancer and prostate cancer [106, 107]. A phase II trial (NCT03513952) treating locally advanced, inoperable, and metastatic urothelial carcinoma with combination therapy consisting of glycosylated recombinant human interleukin-7 (CYT107) and atezolizumab (PD-L1 blockade) is currently underway (Table 1). Lastly, IL-12 may also hold promise in the treatment of urothelial carcinoma and has been incorporated into a novel non-viral gene-based therapy (EG-70) encoding two agonsists of retinoic acid-inducible gene I (RIG-I), which induce production of type I interferons and stimulate innate immunity [108, 109]. A combined phase I and II trial (NCT04752722) studying EG-70 is ongoing for BCG-unresponsive NMIBC patients.
CAR-expressing NK cells
Transfer of unmodified NK cells as a cancer therapeutic has demonstrated positive outcomes primarily in hematologic malignancies but limited utility in solid tumors, highlighting the need for additional strategies [110–113]. One viable approach is recombinant chimeric antigen receptor (CAR) NK cells. Like CAR-expressing T cells, CAR-expressing NK cells contain domains for antigen-binding, signaling (CD3ζ chain or FcɛRγ chain), and one or more co-stimulatory molecules (e.g., DNAM-1 or CD137). Antigen specificity allows CAR-expressing NK cells to recognize tumor cells, leading to NK cell activation and tumor-directed cytotoxicity [46, 114]. Compared to CAR-expressing T cells, CAR-expressing NK cells may offer important advantages, including decreased risk of autoimmunity and malignant transformation due to the short-lived nature of NK cells, a safer cytokine profile and avoidance of cytokine release syndrome (CRS), and the ability to still be effective in the setting of tumor antigen escape [46]. CAR-expressing NK cells also offer the potential to be “off-the-shelf”, as they can be produced from allogeneic sources or from the transformed NK-92 cell line without full human leukocyte antigen (HLA) matching, thereby greatly reducing the time for manufacturing from diagnosis to therapy[115].
Current clinical trials on the efficacy of CAR-expressing NK cells are primarily done in hematologic malignancies. FT596 is an off-the-shelf anti-CD19 CAR-NK cell derived from pluripotent stem cells undergoing phase I study (NCT04245722) in patients with relapsed or refractory B cell lymphoma or chronic lymphocytic leukemia (Table 1) [116]. Seven of 11 patients showed complete response, while two cases of CRS but no graft versus host disease (GVHD) or immune effector cell-associated neurotoxicity syndrome were observed [116]. In another phase I/II study (NCT03056339) of patients with relapsed or refractory B cell lymphoma or leukemia, administration of anti-CD19 CAR-NK cells from HLA-mismatched cord blood led to complete remission and objective response rate of 64% and 73%, respectively, without significant toxicity [117]. Additionally, there are several other phase I studies currently in progress, including the evaluation of anti-CD19 CAR-NK cells in refractory B cell non-Hodgkin lymphoma (NCT04887012), NKG2DL-specific CAR-NK cells in the treatment of recurrent or refractory acute myeloid leukemia (AML) (NCT05247957), and anti-CD33/CLL1 CAR-NK cells in AML patients (NCT05215015) (Table 1). Few trials are investigating CAR-NK cells in the context of solid tumors. These studies include the targeting of prostate-specific membrane antigen in prostate cancer (NCT03692663), mesothelin in ovarian cancer (NCT03692637), HER2 in glioblastoma (NCT03383978), and MUC1 (NCT02839954), NKG2D-L (NCT03415100), and ROBO1 (NCT03940820) in multiple solid tumors including specifically pancreatic cancer (NCT03941457) [114]. Additionally, CD70 has emerged as a potential target for solid tumors as CD70-targeted CAR T cells (named CTX130) are showing promising preliminary efficacy in relapsed/refractory clear cell renal cell carcinoma (ccRCC) (NCT04438083) [118]. In a phase 1 dose escalation trial among 13 treated patients that were evaluated for efficacy.
Future perspectives
Ultimately, the activity of NK cells is dependent on the sum of their activating and inhibitory signals. NK cells perform four major antitumor functions in the TME, including 1. direct lysis of tumor cells via release of preformed, perforins and cytolytic granules as well as expression of the two death-inducing ligands, TNF-related apoptosis-inducing ligand (TRAIL) and Fas ligand (FasL), 2. release of inflammatory cytokines such as IFN-γ and TNF-α, 3. ADCC, and 4. DC recruitment and activation through XCL1, Flt3-L, and GM-CSF [119]. While immune checkpoint blockade is effective at enhancing NK cell function in the TME for a minority of treated patients, it is only one of several available therapeutic modalities. Therefore, the future antitumor crusade requires not only the attenuation of inhibitory signals, but also direct engagement of NK cell activation to utilize NK cells to their fullest potential. Looking to the future, engineered viral vectors, small molecule IMiDs, NK agonist antibodies, and interleukins have demonstrated great potential in addressing this need, and there is great potential in the development of autologous and allogeneic CAR-engineered NK cell products to offer an effective, “off-the-shelf” solution to enhance T cell-targeted immunotherapies. Currently, CAR-NK cells have been primarily tested for their direct antitumor cytolytic functions. It will be important for future studies to appreciate the many ‘helper’ roles that NK cells elicit in shaping their microenvironments and boosting adaptive immunity.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgements.
FUNDING
A.H.: Exelixis, AZ; M.G.: BMS, Novartis, Dendreon, AZ, Merck, Genentech N.B.: Regeneron, Harbor Biomedical, DC Prime, and Dragonfly Therapeutics.
AUTHOR CONTRIBUTIONS
Y.A.W. and A.H. performed the review. Y.A.W. and A.H. wrote the manuscript. D.R., C.B., M.G., N.B. and J.P.S. provided intellectual input.
ETHICAL CONSIDERATIONS
As a review of the literature, and as no animal or human research was involved, our study is exempt from any requirement for Institutional Review Board approval.
CONFLICT OF INTEREST
A.H. is on the advisory boards of HTG Molecular Diagnostics and UroGen. J.P.S. is on the advisory boards of UroGen, Janssen, and Pacific Edge. M.G. is on the advisory board or consultant for Bristol Myers Squibb, Merck, Genentech, AstraZeneca, Pfizer, EMD Serono, SeaGen, Janssen, Numab, Dragonfly, GlaxoSmithKline, Basilea, UroGen, Rappta Therapeutics, Alligator, Silverback, Fujifilm, Curis. N.B. is an extramural member of the Parker Institute for Cancer Immunotherapy; and is on the advisory boards of Neon Therapeutics, Novartis, Avidea, Boehringer Ingelheim, Rome Therapeutics, Rubius Therapeutics, Roswell Park Comprehensive Cancer Center, BreakBio, Carisma Therapeutics, CureVac, Genotwin, BioNTech, Gilead Therapeutics, Tempest Therapeutics, and the Cancer Research Institute.
M.G. and J.P.S. are Editorial Board members of this journal, but were not involved in the peer-review process nor had access to any information regarding its peer-review.