
Abstract
Bladder cancer incidence is drastically higher in males than females across geographical, racial, and socioeconomic strata. Despite potential differences in tumor biology, however, male and female bladder cancer patients are still clinically managed in highly similar ways. While sex hormones and sex chromosomes have been shown to promote observed sex differences, a more complex story lies beneath these evident sex-biasing factors than previously appreciated. Advances in genomic technology have spurred numerous preclinical studies characterizing elusive sex-biasing factors such as epigenetics, X chromosome inactivation escape genes, single nucleotide polymorphism, transcription regulation, metabolism, immunity, and many more. Sex-biasing effects, if properly understood, can be leveraged by future efforts in precision medicine based on a patient’s biological sex. In this review, we will highlight key findings from the last half century that demystify the intricate ways in which sex-specific biology contribute to differences in pathogenesis as well as discuss future research directions.
INTRODUCTION
Bladder cancer (BC) is one of the top non-reproductive cancers exhibiting stark male and female differences: men experience greater incidence by 3–5 times and females are more often diagnosed with advanced disease [1]. Although these differences have been well-documented over the last half century, underlying causes remain obscure.
Sexual dimorphism in a non-pathological setting was once thought to occur after the development of the gonads and subsequent secretion of sex hormones. This framework of understanding is incomplete and consequently led many to heavily emphasize sex hormones as the cause for male and female differences in diseases. Now, we have a deeper appreciation that the baseline unequal chromosomal distribution between biological sexes— such as evidently illustrated by the X and Y sex chromosomes— have both independent and dependent actions with sex hormones to drive sex differences in normal and pathological states [2]. In this review, we will refer to environmental factors, sex hormones, and sex chromosomes as evident sex-biasing factors due to their clear sex-biasing roles (Fig. 1).
Emerging evidence suggests that beyond evident sex-biasing factors, there are elusive sex-biasing effects— such as those from genetic imbalances and epigenetics— that were previously underappreciated due the complexity of their effects and difficulty to experimentally isolate (Fig. 1). In 2018, we discovered sex-specific epigenetics, which we will refer to as the “sex epigenome”, reducing BC risk among individuals with the XX chromosome complement [3]. The advent of cancer genomics such as The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) [4] has also catalyzed a plethora of studies elucidating additional molecular and genetic differences between sexes [5–9].
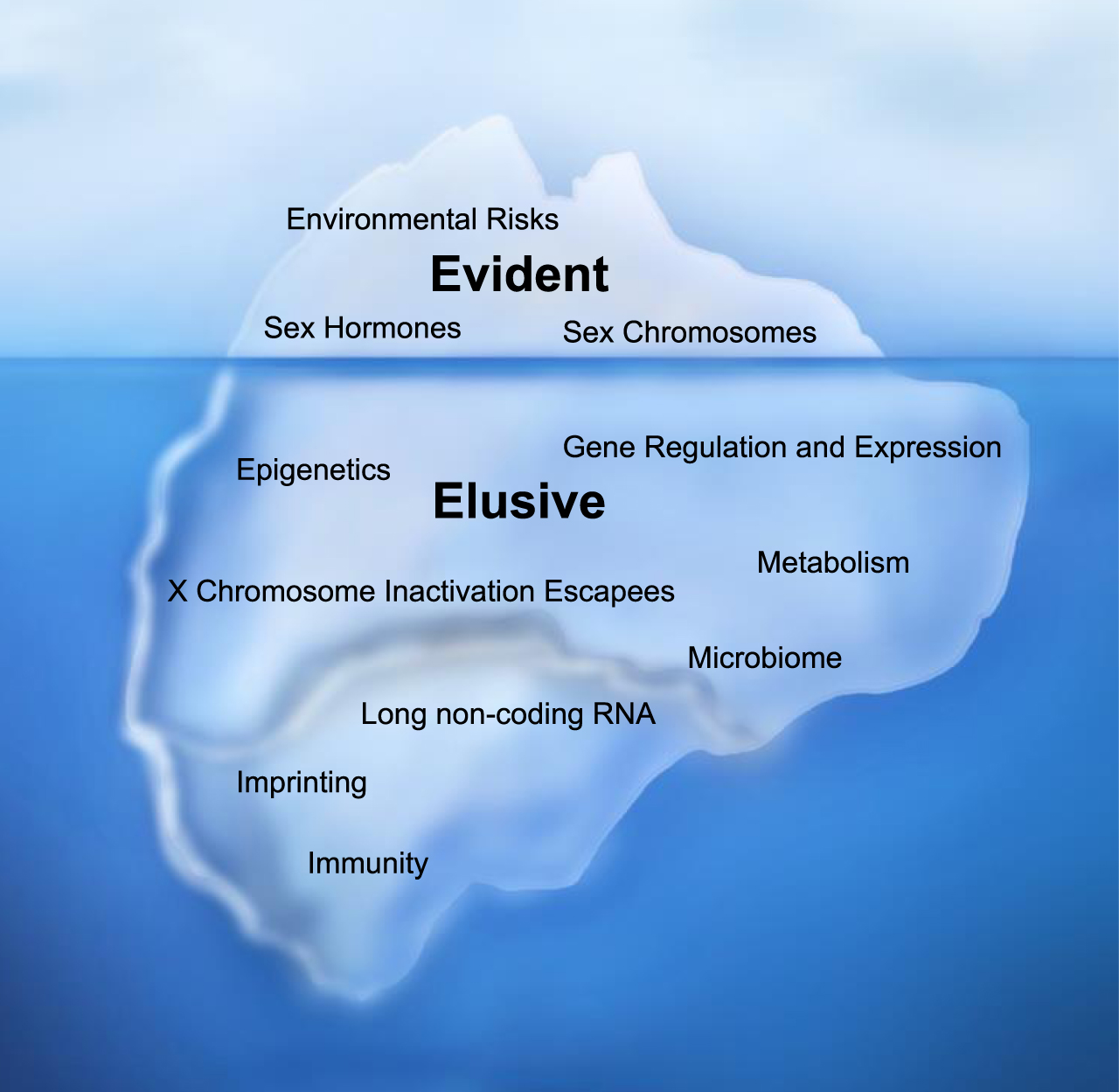
Sex-Biasing Factors of Bladder Cancer. Evident sex-biasing factors differentiating male and female bladder cancer (BC) incidence such as environmental risks, sex hormones, and sex chromosomes have historically been emphasized but do not accurately illustrate the full scope of factors driving sex differences. Many more hidden yet important factors, which we describe as elusive factors, may contribute to higher male BC incidence; they include: epigenetics, gene regulation and expression, X chromosome inactivation escape genes, metabolism, the microbiome, long non-coding RNAs, imprinting, and immunity.
Despite the field’s substantial preclinical progress, male and female BC patients are still clinically managed in highly similar ways, aside from anatomical differences [10]. High recurrence, resistance to standard therapies, and the financial burden of BC calls for an overdue clinical shift towards precision medicine based on biological sex to improve sex-specific survival and lower treatment costs. In this review, we will highlight both the evident and elusive sex-biasing factors of BC to provide an overview of the complex interplay of variables driving sex differences in BC and propose future research directions (Table 1). Properly understanding both evident and elusive sex-biasing effects can expedite future design of sex-specific BC screening, prevention, and therapeutics to mitigate existing sex disparities.
Summary of sex-biasing factors and corresponding publications
Note: TBD, to be determined; AR, androgen receptor; ER, estrogen receptor; LOY, loss of Y chromosome; KDM6A, lysine demethylase 6A; PRC2, methyltransferase of polycomb repressive complex 2; EZH2, enhancer of zeste homolog 2; COMPASS, complex of proteins associated with Set1; TOP2B, DNA topoisomerase 2 beta; UGT, UDP-glucuronosyltransferases; ROS, reactive oxygen species.
EVIDENT SEX-BIASING FACTORS
Behavioral and Environmental Factors
Tobacco smoke is a major risk factor for BC [11]. Unequal smoking patterns between males and females provides a reasonable explanation for observed sex disparities. Yet, prevailing differences in BC incidence among males and females with similar smoking levels confirm the hypothesis that smoking itself is not the only cause of sex disparities [12–14].
Due to the historic workplace of men, higher rates of occupational exposures in men compared to women— especially for workers near aromatic amines, leather, paint, industrial machines, and aluminum— were once deemed as likely factors driving higher BC incidence in men. In 1990, however, the National Bladder Cancer Study by Hartge et al., showed that environmental exposure and cigarette smoking did not fully explain sex differences in BC [14].
For decades, many believed that environmental factors explained male and female differences in BC incidence and prognosis. However, these aforementioned studies suggested otherwise— marking the beginning of a turn away from relegating environmental factors as the sole culprit to truly investigating sex as a biological variable as a viable factor of sex bias in bladder tumor development and therapy.
Sex Hormones
Androgens and Androgen Receptors (AR)
Sex steroid hormones and their respective receptors in BC have been studied for nearly half a century. In 1972, androgens were first identified as a BC risk factor when castrated male mice were better protected against bladder carcinogenesis induced by N-butyl-N-(4-hydroxybutyl)nitrosamine (BBN) than uncastrated males, and female mice treated with testosterone had increased BC risk compared to control female mice [15]. Preclinical studies further suggested that androgens induce while estrogens suppress carcinogen-induced BC development in rats [16]; clinical observation of men having decreased BC mortality after treatment with 5α-reductase inhibitors or dihydrotestosterone (DHT) blockers further solidified the relevance of androgens [17]. Ultimately, these studies were hypotheses-generating, as it remained unclear how exactly androgens initiated or potentiated BC. The next round of breakthrough studies came from molecular characterization of the androgen receptor (AR).
Using patient samples, Laor et al., observed greater levels of AR in tumor tissue compared to normal tissue, and females had relatively lower levels than males [18]. High grade tumors— although intuitively expected to exhibit increased AR— paradoxically had less AR than low grade tumors. It should be noted that other studies showed conflicting results, and this could be due to a lack of standardized staining and scoring [19, 20]. Usage of genetically engineered models (GEMs) subsequently led to a series of insightful studies. Mice with AR gene knockout (ARKO), either germline [21] or urothelium-specific knockouts [22], were less susceptible, while mice expressing extra AR (hARGoF) [23] were more susceptible to BC induced by a bladder-specific carcinogen. The genetic evidence convincingly demonstrated the essential role(s) of AR in promoting bladder tumorigenesis. Findings from Miyamoto et al., further implicated how AR and androgens did not completely rely on one another to mediate their cancer driving effects [21]. For example, DHT treated and untreated male ARKO mice developed bladder carcinoma at rates of 25% and 0%, respectively, suggesting the function of androgens via a non-AR pathway. Consistent with these implications, Chang and colleagues recently reported a membrane androgen receptor (mAR-SLC39A9) that functions through a noncanonical AR pathway Gαi/MAPK/MM9 to promote bladder cancer [24].
Mechanistic studies on AR have led to the identification of downstream target genes and molecular pathways. Initial characterizations of AR knockout mice suggested that tumor promoting activity of AR depends on the p53-PCNA DNA damage repair pathway [22]. Indeed, somatic loss-of-function of p53 tumor suppressor gene is frequently observed in BC [25, 26]. To validate whether AR promoting bladder tumorigenesis required the p53 pathway, Hsu et al used simian vacuolating virus 40 T (SV40T) to inactivate tumor suppressors p53 and Rb in mouse lines with or without urothelial AR; results showing no difference in bladder tumor initiation between the two groups supported the notion that AR bladder tumor initiating activity requires modulation of p53. The activity of AR in inducing BC cell proliferation in vitro [21, 27] also appears to be dependent on CD24, a glycosyl phosphatidylinositol-linked sialoglycoprotein— a direct downstream target of AR [27]. Interestingly, inactivation of CD24 is protective against BBN-induced BC in male but not in female mice, suggesting that the sex-biasing activity of AR is in part mediated by CD24 in vivo [27]. Moreover, Theodorescu and colleagues demonstrated that AR directly represses gene transcription of CD44 [28], the receptor for hyaluronic acid— a biomarker and potent driver of progressive disease in multiple tumor types, suggesting that AR may function in a stage dependent manner in promoting bladder tumor initiation but suppressing tumor progression.
Ectopic activation of the Wnt/ β-Catenin signaling pathway in the bladder urothelium induces luminal bladder tumor in mice [29]. Like the BBN-induced bladder carcinogenesis model, this GEM model of BC also displays a striking sex difference: the incidence is 45% in males vs. 3% in females when the Wnt/ β-Catenin pathway is constitutively activated in the bladder urothelium. Mechanistically, constitutive β-Catenin activation induces nuclear translocation of AR while conversely, activation of the AR pathway potentiates the Wnt/ β-Catenin signaling in bladder urothelial cells. Similar genetic interactions between androgen and Wnt/ β-Catenin signaling pathways are also found during the masculinization of external genitalia [30]. Together, these observations suggest that synergism between the AR and Wnt/ β-Catenin pathways may enhance sex differences in development and diseases.
Studies have also suggested the roles of androgens in dampening the host immunity against cancer, resulting in increased male cancer incidence [31] and mortality [32]. For example, androgens have been shown to be immunosuppressive by inhibiting the function of macrophages, NK cells and T cells [33]. When exposed to persistent antigen stimulation and hostile tumor microenvironment (TME) for tumor-infiltrating T cells, tumor-specific T cells are known to undergo programmatic dysfunction and exhaustion with diminished ability to mount effector function against cancer [34]. The mechanism of molecular programming of T cell exhaustion is not entirely clear, but the process involves key transcriptional and epigenetic changes centered around important transcription factors including Tcf7 (encoding Tcf1) and TOX [35, 36]. Using single cell RNA sequencing coupled by transcription studies, we recently discovered that androgens can promote CD8+ T cell exhaustion in the TME by directly transactivating Tcf7 [37].
Overall, this field has taken significant strides to characterize androgens/AR pathway in male BC incidence and progression. However, key questions remain: Are androgens and AR solely responsible for sex differences in BC development? Will targeting these pathways address sex differences more than existing therapies? While clinical trials (e.g., NCT02605863, NCT01234519, NCT02788201, NCT02300610)— including those testing the efficacy of existing hormone therapy for prostate cancer on recurrent NMIBC or advanced BC— had been initiated to test preclinical findings, trials consisted of small cohorts and in some cases were suspended due to insufficient enrollment. Meaningful clinical application would necessitate clinical trials with a larger patient cohort that includes both males and females.
Estrogens, Estrogen Receptors (ERα and ERβ), and Progesterone
Menopause provides a natural but indirect way to observe the effects of estrogen depletion in women, given that effects are confounded by age. In a 2006 prospective study, post-menopausal women had increased BC risk and early age menopausal onset yielded even greater risk, suggesting the protective effects of estrogens [38]. These findings are preceded by Bertram and Craig’s initial observation of estrogen’s protective effects in female mice contrasted with testosterone’s effects to increase BC risk in male mice [15]. As straightforward as this seems, a closer look at the relationships between estrogens, estrogen receptors, and progesterone reveals a more complex story.
Estrogens can function through two canonical nuclear receptors— ERα and ERβ— each responsible for potentially distinct functions, especially depending on tissue type. In rat and mice BC, ERα was inconsistently detected [39, 40]. Thus, unlike breast cancer, ERα did not seem to have prognostic value. However, in 2014, Hsu et al., showed that ER α knockout mice developed tumors faster than females with ER α, suggesting ERα’s inhibitory role in tumorigenesis [41]. ERβ, on the other hand, is expressed in human tumor tissues and increases with higher stage and grade BC, suggesting its role in tumor progression and metastasis [42]. In female mice, depleting ERβ slowed down tumor growth [43]. Thus, ERα may be the predominant driver of tumor initiation and ERβ of tumor progression. It has also been shown that Tamoxifen— a selective ER modulator (SERM) that inhibits ER activity— administered before, during, or after BBN treatment conferred protection from urothelial carcinogenesis in female mice [44]. Thus, existing studies imply: (1) ERα and ERβ serve different functions during BC initiation and progression, (2) Tamoxifen or SERM could act as a chemoprotective agent against BC, and (3) future studies should specify tumor type and stage when studying the mechanisms of ERα, ERβ, and SERMs.
Progesterone may also play a role. Parous female mice— those who gave birth and thus experienced changes in progesterone— developed smaller bladder tumors than nulliparous mice [45]. In humans, multiparous females had decreased BC risk [46, 47]. Thus far, there have not been follow-up studies to investigate the underlying mechanisms behind these observations; it would be interesting to see if progesterone mechanistically acts independently or interactively with ER receptors to affect BC risk in females. Finally, it is important to note that although males have lower levels of estrogens and progesterone, the actions of predominantly female sex hormones should not be ignored in males and vice versa.
Ultimately, sex hormones can only be partially responsible for BC sex disparities. Given that the median age of BC diagnosis is 73— an age with decreased sex hormone levels— how can sex hormones be the sole responsible player? Other critical sex-biasing factors must also be at play.
Sex Chromosomes
In mammals, there is an unmistakable genetic inequality between sexes: males have one copy of the X and Y sex chromosomes, while females have two copies of the X chromosome. Activity of Sex-determining Region Y (Sry) encoded by the Y chromosome initiates differentiation of the gonads into the testes instead of ovaries and androgens secreted by the testes further induce male–specific sex differentiation. Because of the dominant roles of gonadal hormones, sex chromosomes have historically been overlooked for their potential function to induce male and female BC differences and has, only in more recent years, been studied for its unique sex-biasing effects. A turning point began after the 2008 epidemiological study revealed how Turner syndrome patients— phenotypic females completely or partially missing one copy of the X chromosome (i.e., XO)— had higher BC risk than XX females [48]. In the same vein, Klinefelter’s patients— phenotypic males with one or more extra X chromosome(s) (e.g., XXY)— had lower risk of solid tumors than XY males [49]. While these studies suggested the potential roles of sex chromosomes, they were correlative and could not definitively conclude the independent effects of the X and Y chromosome because confounding factors such as chronic urinary tract infections, altered levels of sex hormones, or other pre-existing comorbidities were not accounted for.
To directly examine the role of sex chromosomes (XX vs XY) in BC risk, we used age-matched “four-core genotype (FCG)” mice, which consist of four sex types: two testes-bearing types with either XX or XY chromosomes and two ovaries-bearing types with either XX or XY chromosomes [3, 37]. The FCG technology uncouples the sex chromosome effect (SCE) and gonadal hormone effect (GHE), thereby enabling quantitative evaluation of independent as well as interactive effects of these central sex-biasing variables [50]. We treated FCG mice with BBN and monitored BC development and overall survival [3]. Cox proportional analysis confirmed the independent sex-biasing effects of the testis or androgen hormones with a hazard ratio of 4.714 (vs ovary, 95% CI = 2.77–8.28). In addition, it also uncovered that the sex chromosome complement is a sex-biased risk factor independent to the gonadal hormones (hazard ratio of 2.549, 95% CI = 1.55–4.28). Surprisingly, there was an interactive effect between GHE and SCE because the combined hazard ratios from the sex hormone and sex chromosome effects was 12.39 (95% CI = 5.54–31.63)— equivalent to the product rather than sum of 2.549 and 4.714 (Table 2). These findings suggested, for the first time, that SCE and GHE may be synergistically interacting to amplify sex differences in BC (Fig. 2).
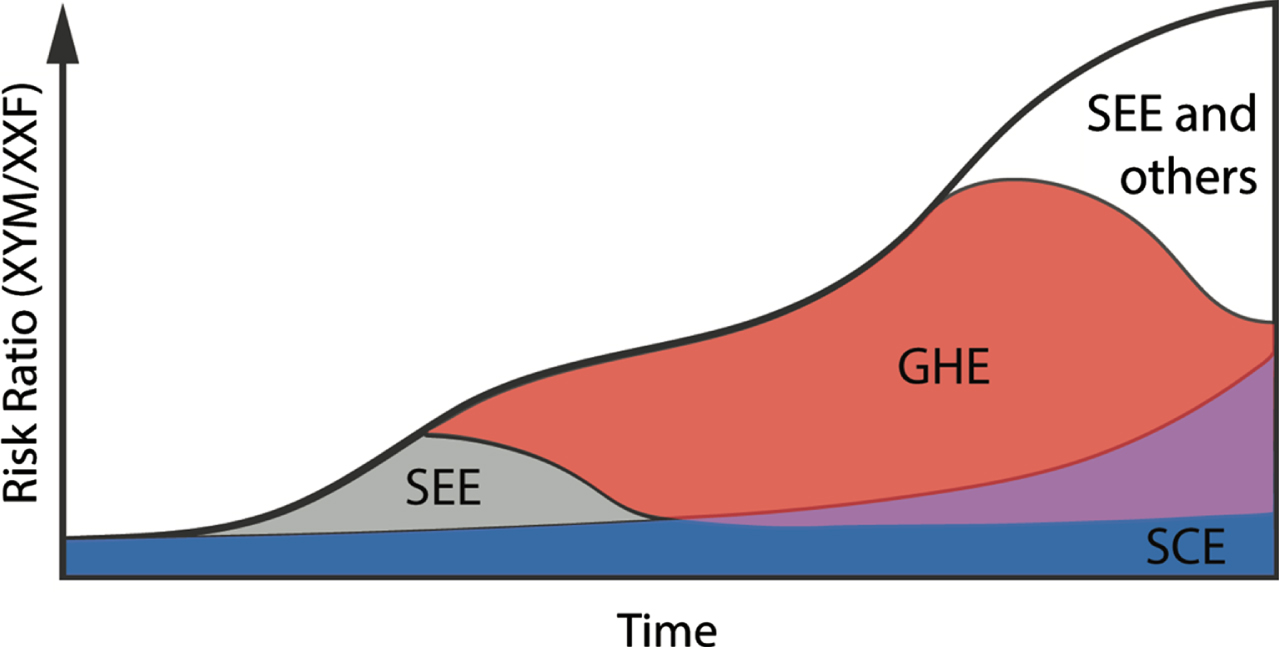
Independent and Interactive Effects of Sex-biasing Factors. A proposed mechanism on how sex chromosome effects (SCE), gonadal hormone effects (GHE), the sex epigenome effect (SEE), and other sex biasing factors independently and dependently contribute to drive higher incidence of bladder cancer in males with time. SEE may interact with SCE and GHE to increase bladder cancer risk in males as patients age. Blue, SCE; gray, SEE; red, GHE; purple, interaction between SCE and GHE; white, SEE and other sex biasing effects.
Independent and cooperative effects of sex chromosomes and gonadal hormones to drive sex differences in BC risk
SCE, sex chromosome effect; GHE, gonadal hormone effect; HR, hazard ratio.
Loss of Y chromosome (LOY)— shown to be induced by smoking— frequently occurs in males with BC [12, 51–54], but its lack of association to tumor stage or patient survival has caused many to question its clinical importance [12, 55]. However, Forsberg et al., showed that age-related LOY in men resulted in a median shorter lifespan of 5.5 years compared to men without LOY, suggesting that Y chromosomal effects on cancer risk cannot be forgone [56]. Therefore, it is possible that the Y chromosome suppresses BC development and LOY increases BC risk. Hence, presence of the Y chromosome is very unlikely to be a reason of increased BC risk in males. One potential role of the Y chromosome may be relegated to epigenetic regulation which will be discussed later.
ELUSIVE SEX-BIASING FACTORS
Genetic Imbalance
X Chromosome Inactivation (XCI) Escapees
One copy of the X chromosome in XX female cells is normally inactivated in a random manner through a process called X Chromosome Inactivation (XCI). This dosage compensation mechanism ensures that X-linked genes are expressed at comparable levels between XX and XY cells. However, XCI is an imperfect process— a fraction of genes escape XCI, leading to higher levels of XCI escapees expressed in XX females than in XY males. In humans, nearly 23% of X-linked genes escape XCI [57], which supports the hypothesis that XCI escapees can contribute to sex differences in cancer. Because females effectively have two functional copies of these escapees while males have only one copy, losing one functional copy in males is sufficient to cause a phenotypic change while females will still have one functional copy to compensate the loss. Based on this premise, Lane and colleagues analyze paired tumor–germline exome sequencing data from 4,126 patients across 21 tumor types from TCGA and Broad Institute data sets and identified that loss-of-function mutations of six X-chromosome genes (ATRX, CNKSR2, DDX3X, KDM5C, KDM6A, and MAGEC3) are more frequently found in males than females [5]. Of the 6 named genes, only KDM6A exhibited relevance in affecting sex differences in bladder cancer, which we will further discuss in the section titled Epigenetics: The Sex Epigeneome.
Sexually Dimorphic Genetic Architecture
It is worth noting that in addition to X and Y sex chromosomes, autosomes may also contribute to sex differences in cancers. By interrogating the ICGC/TCGA data, Li et al. showed that somatic mutation frequency of an autosomal gene CTNNB1 in liver hepatocellular cancer is significantly higher in males than in females, implying that there are sex differences in oncogenic mutational processes beyond the mutational differences of X-chromosome-encoded genes [7, 8]. Genetic variations can also occur in non-coding regions to influence gene regulation and expression. Collectively, these genetic variations, linked to observed quantitative differences in gene expression, can be referred to as an individual’s genetic architecture. Here, we will discuss how differences in genetic architecture contribute to sexual dimorphism.
Advancement of genomic techniques and curation of large genomic databases, such as the Genotype-Tissue Expression (GTEx) project [58], has made studying sex differences of genetic architecture— down to the level of a single nucleotide— feasible. Using GTEx data, Lopes-Ramos et al. interestingly observed more sex differences on the level of transcription regulation than gene expression in a tissue specific manner: 87% of genes were differentially targeted, meaning they experienced different regulatory pressures, but 70% of these differentially targeted genes, did not show differential expression [59]. This means that certain phenotypes may manifest in a latent manner as time, age and stress differentially impact male and female specific transcription factors. Meaning, at any given time point, the potential for differential expression may be higher than observed due to differences in regulation hidden beneath more uniform expression. Oliva et al. extended upon these finding by analyzing the cis sex-biased expression quantitative trait loci (eQTLs) and performed allelic expression analysis between males and females [60]. Their findings reiterated the concept that a seemingly similar phenotype between males and females is supported by a diverse set of sex-specific checks and balances. They identified sex-biased genes in pathways such as epigenetic methylation and xenobiotic metabolism including CYP450 genes.
One limitation of these recent studies using the GTEx dataset is the lower population data for bladder, endocervix, ectocervix, fallopian tube, renal medulla than the rest of the tissue types (n < 25 vs. n = 73–670), resulting in the exclusion of these five tissues from the analyses. In general, this avenue of research has not yet been extensively applied to study sex differences in BC. Previous efforts of genome-wide association studies (GWAS) have identified several loci that are tightly associated with BC risk [61–67]. We believe that analyzing gene regulation such as eQTLs could be insightful in explaining male and female differences in BC incidence and mortality by revealing the sex-specific variability in response to the environmental signals and pathological states.
Sex-specific selection constraints may be another potential mechanism contributing to a sex-specific genetic architecture. In two different studies, Gershoni et al., found that both men and women had reduced selection pressures on sex-specific genes compared to non sex-specific genes [68, 69]. Moreover, the magnitude of decreased selection was greater in men than women [69]. They postulate that the reduced negative selection in male-biased genes may contribute to a greater accumulation of deleterious mutations in men and in turn yield different patterns of disease incidence. Whether this mechanism is relevant in explaining BC sex differences is not yet known and should be further explored.
Epigenetics: The Sex Epigenome
Epigenetic regulation lies at the heart of genetic modulation and phenotypic diversity. The epigenome plays a central role in cancers, and epigenetic regulators are most frequently mutated in BC than in other solid tumors [25, 70–72]. While barriers studying this complex system may have dampened rapid initial progress, its stable traction in recent years along with the emerging concept of sex-specific epigenetics— which we will collectively refer to as the sex epigenome— brings a promising outlook to translational application.
Modifications such as methylation and acetylation of DNA and histones (as well as removal of the modifications) define the simple yet ingenious way for cells to selectively silence or promote expression of specific genes to adapt to biological and environmental changes. Abnormal or pathological states often hijack or disturb this intricate epigenetic system. We define the sex epigenome as sex-specific differences in post-translational modification and chromatin organization that functionally influence gene expression and/or response to the environment [3]. The sex epigenome is likely influenced by effects from sex hormones, sex chromosomes, and additional sex-biasing factors (Fig. 3).
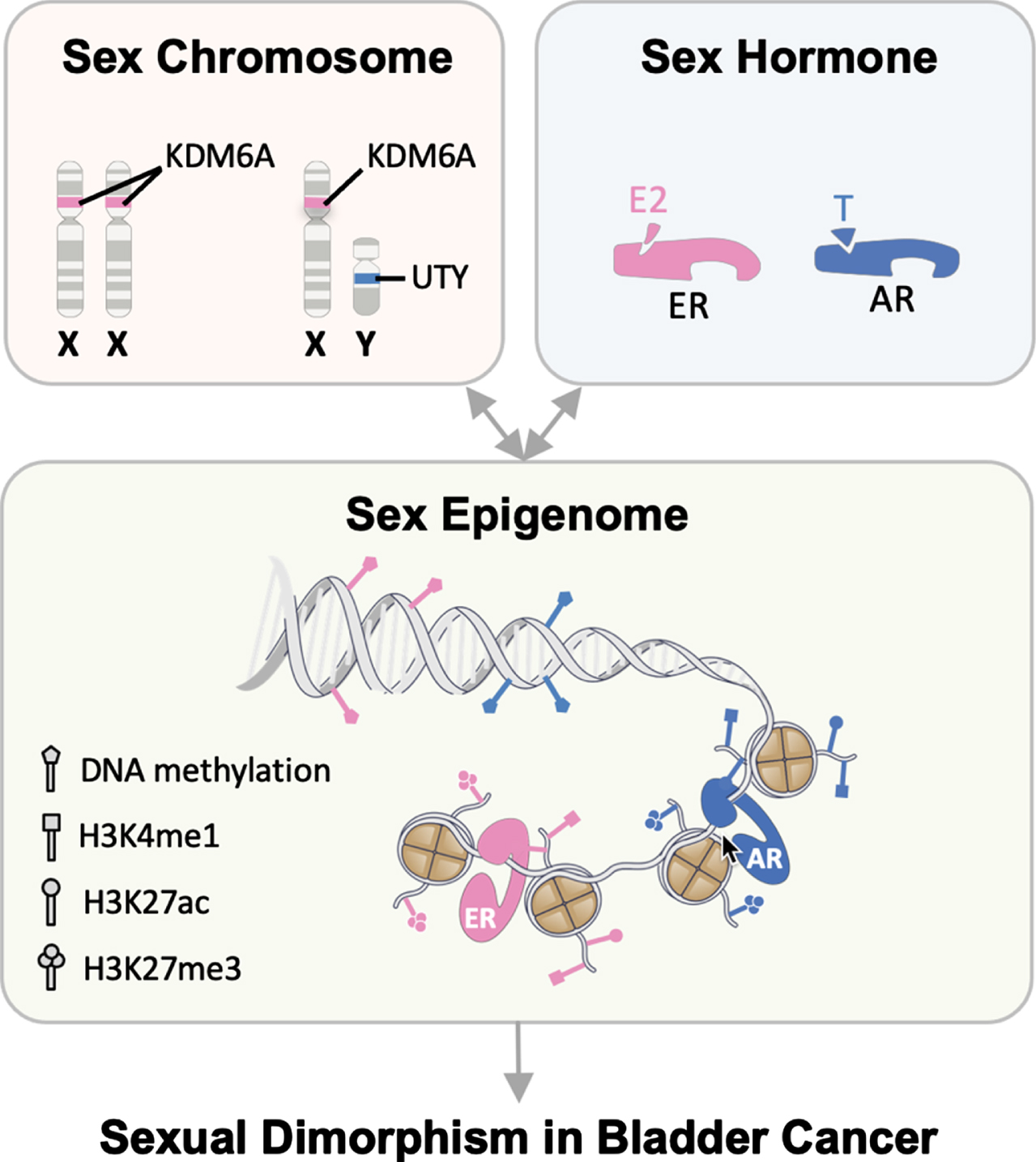
Cumulative and interactive effects from sex chromosomes, sex hormones, and the sex epigenome result in sexual dimorphism in BC. Sex-specific differences in post-translational modifications and chromatin organizations, also known as the sex epigenome, functionally influence gene expression and/or response to the environment. We propose a novel framework to understand the drivers of sex differences in BC: the sex epigenome acts as a universal platform where the effects of sex chromosomes and sex hormones converge to result in the observed sex bias in BC.
Lysine demethylase 6A (KDM6A)— also known as ubiquitously transcribed tetratricopeptide repeat on chromosome X (UTX)— is commonly mutated in BC; its mutated form loses the normal histone demethylase function on tri-methylated histone H3 at lysine 27 (H3K27me3), making H3K27 available for acetylation. In the BBN model of bladder carcinogenesis, XY males are 12.39 times more likely than XX females to develop and die from BC (Table 3). In 2018, we found that urothelium-specific deletion of KDM6A significantly increased BC risk of females but not male mice [3]. KDM6A deletion alone effectively reduced the male-to-female BC risk ratio by more than five-fold (12.39 vs 2.349), suggesting that KDM6A functions as a prototypical female-biased tumor suppressor. Transient expression of KDM6A in a human BC cell line (UM-UC-13) elicited tumor suppressing results through its demethylase activity, suggesting a cell’s ability to retain epigenetic memory from even a temporary expression of KDM6A. We verified these findings using transcriptomic analysis of patient data from TCGA project. Mechanistically, KDM6A promoted expression of known canonical Tp53 gene targets (CDKN1A and PERP) [3, 73]. KDM6A conditional knockout male mice did not have worse survival compared to female counterparts. This may be due to the Y chromosome encoding for UTY, a paralog of KDM6A; UTY on the Y chromosome may compensate the loss or mutation of KDM6A on the X chromosome. While UTY has been shown to have no or little detectable demethylase activity [74–79], its demethylase independent function could suppress BC development.
Kdm6a is a primary sex-biasing factor responsible for the protective effects of the X chromosome in BC
N, sample size; M, male; F, female; HR, hazard ratio.
Polycomb repressive complex 2 (PRC2) is responsible for chromatin compaction and gene expression. Enhancer of zeste homolog 2 (EZH2), the methyltransferase of PRC2, has an antagonistic relationship with KDM6A to control the methylation status of H3K27 in the bladder urothelium [80]. In a preclinical study, it has been shown that recovering KDM6A demethylase function indirectly through EZH2 inhibitors is therapeutically effective [81]. Because KDM6A mutations are more common in women with non-muscle invasive BC [72], future studies should consider the viability of targeting KDM6A related pathways in female BC patients with EZH2 inhibitors.
Independent from its demethylase action, KDM6A operates with the complex of proteins associated with Set1 (COMPASS) family, specifically two proteins called mixed lineage leukemia 3 and 4 (MLL3/KMT2C and MLL4/KMT2D), to mediate methyltransferase activity of H3K4me1 [82–88]. Drawing from current evidence, KDM6A serves to attenuate tumorigenesis by two different histone modifying mechanisms: (1) antagonizing the PRC2-dependent transcription repression via H3K27 tri-methylation and (2) promoting the COMPASS-dependent transcription activation via H3K4 mono-methylation [89]. While the potential sex-biasing roles of PRC2 and COMPASS complexes have not yet been proven in BC, their dynamic and diverse roles in development and cancer emphasizes the potential to leverage their actions to mitigate sex differences in BC.
DNA topoisomerase 2 beta (TOP2B) showed male-biased DNA methylation in BC patients, and Valrubicin— an analog of doxorubicin used for BCG refractory patients— is designed to antagonize topoisomerase activity [6]. According to the 2021 National Comprehensive Cancer Network (NCCN), current Valrubicin recommendations are not specific to a patient’s biological sex. Thus, more research is warranted to elucidate how the sex epigenome and sex hormones may differentiate BC patients’ response to Valrubicin.
Piecing together the epigenetic basis for BC sex disparities could reveal foundational mechanisms driving sex differences in BC patients. Thus, a systematic characterization of the sex epigenome in males and females could open many doors for precision medicine in BC. Ultimately, we believe that sex differences may be driven by a coordinated effect between three major players: the sex epigenome, sex hormones, and sex chromosomes and, moreover, interactions among these players may amplify the magnitude of sex differences (Figs. 2 and 3).
Metabolism
Metabolic differences between males and females are well-known, but its role in driving sex differences in cancer— especially in the metabolic reprogramming of cancer cells— has only been a more recent consideration [90]. At baseline, male serum has higher concentrations of carbohydrate and amino acid metabolites [91], while female cells have a predilection for lipid biosynthesis [92]. Rubin et al., posited that male cancers may specifically reprogram carbohydrate and amino acid metabolism and female dominated cancers may selectively reprogram the normal utilization of lipids [90]. It is not yet known whether this effect plays a part in increasing male bladder cancer risk.
Biotransformation of molecules such as hormones, neurotransmitters, drugs, and xenobiotics in relation to environmental exposures, usage, accumulation, and elimination is also an important metabolic process that has shown sex differences. In a case-control study, Zheng et al., observed that a His213 allele of a sulfotransferase gene (SULT1A1) conferred decreased BC risk only in females [93]. Further studies are needed to validate the clinical application of this finding as the female composition of the study was only around 24%.
The UDP-glucuronosyltransferases (UGT)-dependent detoxification pathway is responsible for eliminating xenobiotics and endobiotics [94]. The human UGT loci has been closely linked to BCa risk [61–67, 95–98] and in liver tissue, it has been shown that men have a higher expression of UGT enzyme UGT2B17, suggesting different enzymatic activity to metabolize carcinogens and chemotherapeutics between sexes [99]. Downregulation of UGTs is closely associated with tumor formation in mice [100, 101] and humans [102, 103]. AR represses expression of UGTs in the bladder [103] and the prostate [104], implying a sex-biasing role of the UGT detoxification pathway in BC. Despite the strong association, there is a lack of evidence demonstrating causality. Currently, it is not well-established whether such sex differences persist in normal or cancerous bladder tissue. A notable preclinical barrier hindering current research is due in part to species differences between mouse and human UGT superfamily genes [105]. Humanized mouse models can overcome this barrier to elucidate the roles of UGTs in BC carcinogenesis and potential sex-biasing effects.
Mitochondria, another important player in metabolism, are maternally inherited and exhibit strong sex-specific activity in normal and pathological conditions [106]. One of the outputs of mitochondrial activity is reactive oxygen species (ROS), and it has been suggested that female mitochondria have a better ability to maintain lower ROS levels than male mitochondria in the brain [90]. In some ways, this leads to a more susceptible tumor environment for males. It remains to be shown whether this phenomenon also affects male and female bladders. Numerous factors, including ROS levels, differentially drive mutations in the mitochondrial genome, generating a mosaic landscape even within an individual. Investing into the search for potential links between mitochondrial genome mutations, respiratory capacity, ROS levels, and sex differences in BC risk will be challenging but could prove fruitful.
FUTURE DIRECTIONS AND CONCLUSION
Sex disparities in BC morbidity and mortality are intuitively and clinically well accepted, but the full scope of mechanisms remains unknown. Preclinical publications have illustrated the roles of evident sex-biasing factors such as environmental toxins, androgens, estrogens, their respective receptors, and sex chromosomes (Table 1). But a deeper dive into the biology implies crosstalk between the aforementioned factors with more elusive effects (Figs. 1–3Table 1). Thus, to adequately address the age-old question as to why males have greater BC incidence, we must adopt a framework of understanding that includes sex-biasing factors that have historically been elusive— these include XCI escapees, sex-biased transcriptional regulation, sexually dimorphic gene expression, the sex epigenome, metabolism, etc. With the advancement of genomic technology, numerous sex-biasing factors identified in other diseases remain to be characterized in BC. As mentioned before, androgen–mediated promotion of CD8+ T cell dysfunction contribute to sex differences in BC, highlighting the immunological basis of sex disparities in cancers [37]. The field of sex differences has only begun to scratch the surface of the immunological basis of sex bias in cancer. In addition, age-related pathways, microbiome, genomic stability, imprinting, and long non-coding RNA are all potential avenues that should be further explored.
For the purposes of this review, we painted a clear difference between male and female biology. However, it is important to note that individuals with the same biological sex may exhibit different degrees of a certain sex-specific biological trait; for example, a female biased gene regulator may be present at different levels between two biological females [107]. Thus, overall, in designing future experiments, we believe that a simple comparison between males and females is not adequate. Every study should always: 1) consider the potential spectrum of male and female specific effects, 2) isolate the effects of each sex-biasing factor to eliminate potential confounding effects, 3) measure any potentiating or neutralizing effect one factor has on the other, and 4) adjust for sex specific age-related changes such as declining hormonal levels unique to men and women. Using proven strategies such as the FCG model is one way to address confounding factors.
In conclusion, we highlighted preclinical results from the last decade with the goal to guide and direct future studies to formulate testable hypotheses that properly address how elusive and evident sex-biasing factors promote drastic male and female differences in BC. We hope that by tackling these specific areas of research, we can catalyze clinical improvement for males and females in the prevention, screening, and management of BC.
Footnotes
ACKNOWLEDGMENTS
We would like to thank all the members of the X.L. lab as well as Drs. Seth Lerner and Andrea Apolo for their insightful comments and suggestions.
FUNDING
This work was supported by NIH/NCI (R21CA2 49701, X.L), NIH/NIDDK (U01 DK131377, R01 DK091645, X.L.) and NHLBI (1R01HL136921, X.L.).
AUTHOR CONTRIBUTIONS
CML and XL wrote and edited the manuscript. All authors contributed to the concept of the review.
CONFLICT OF INTERESTS
CML, DT, and XL declare no conflict of interests. ZL a scientific advisory board member for Heat Biologics, Alphamab (chair), Hengenix and Ioknisys. DT is an Editor of this journal but was blinded and not involved in the peer-review process nor had access to any information regarding its peer-review.