
Abstract
Presuming that Alzheimer’s disease (AD) might represent an antagonistic pleiotropic phenomenon derived from the evolvability of multiple amyloidogenic proteins, targeting such proteins simultaneously could enhance therapeutic efficacy. Furthermore, considering that amyloid-β (Aβ) immunotherapies during reproductive life stage might adversely decrease Aβ evolvability in an offspring’s brain, the disease-modifying Aβ immunotherapies should be limited to post-reproductive time in lifespan. Thus, current Aβ immunotherapy strategies should be revised accordingly. Given that the “adiponectin paradox” might underlie both amyloidosis and cognitive dysfunction in aging brain, blocking activin signaling situated downstream of the adiponectin paradox might be an alternative strategy to prevent AD.
Keywords
Despite two decades of Alzheimer’s disease (AD) clinical therapeutics, the prevailing strategy of amyloid-β (Aβ) immunotherapy has never convincingly met all benchmarks for therapeutic efficacy. Furthermore, early Aβ immunotherapy trials employing active recombinant Aβ42 immunization were suspended due to encephalomyelitis [1]. Notably, a dissociation has existed where histologically demonstrated improvement in Aβ neuropathology in AD patients, was paired with a lack of improvement in clinical dementia [2, 3]. Newer passive Aβ immunotherapies in aged patients using monoclonal and polyclonal Aβ antibodies [4] have also shown little clinical benefit (Fig. 1A).
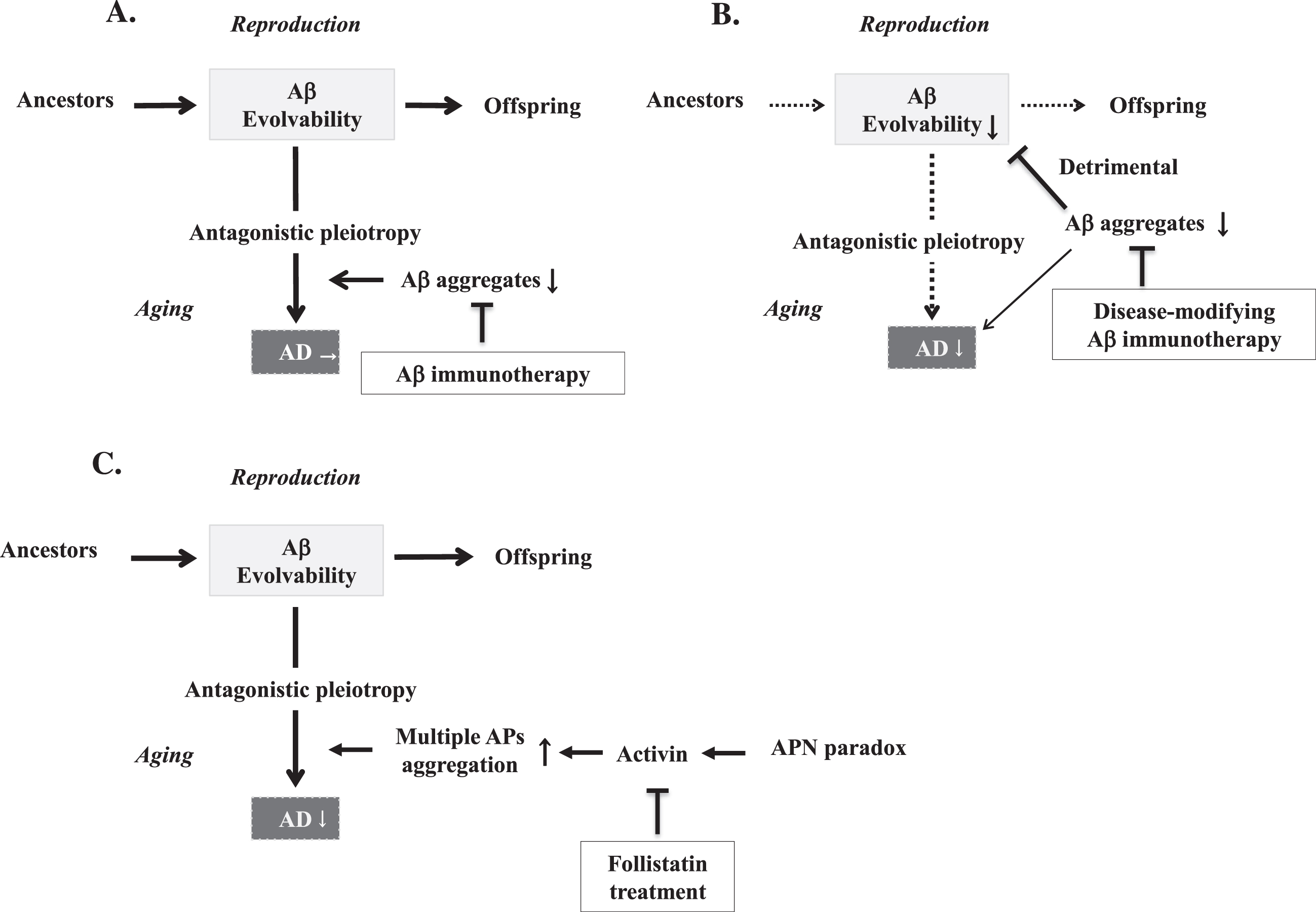
Diagram illustrating the various effects of therapeutic strategies on amyloidogenic evolvability and neurodegenerative disorders. Evolvability in offspring and AD in parental aging are driven by aggregation of APs, and these states are proposed to exist in an antagonistic pleiotropy relationship. A) Conventional active Aβ immunotherapies carried out for AD during aging have proved unsatisfactory and unforeseen side effects. B) Second generation disease-modifying Aβ immunotherapies potently inhibit various stages of Aβ aggregation, resulting in both suppression of Aβ evolvability and AD. Thus, disease-modifying Aβ immunotherapy is beneficial to the older AD patients, but detrimental to their offspring. C) The adiponectin paradox-activin signaling pathway is involved in the regulation of Aβ evolvability, and later during aging, in which activin may promote AD neurodegeneration through the antagonistic pleiotropy mechanism. Therefore, treatment with the activin-binding molecule, follistatin, or analgoues [25, 26], may be effective in preventing activin-induced AD neurodegeneration.
The precise reasons for Aβ immunotherapy failure in AD remain elusive. Central to this remains our incomplete understanding of the normal physiology of amyloidogenic proteins (APs), like Aβ and tau, relevant to neurodegenerative disease. Consequently, we proposed evolvability as a novel physiological function of APs in neurodegeneration [5, 6]. Specifically, as APs consist of intrinsically disordered structures [7] corresponding to diverse brain stressors, their protofibrils might transmit stress information to offspring via germ cells in a prion-like fashion [8]. Supposing that amyloidogenic evolvability might critically influence the success of amyloid immunotherapy, our objective is to re-imagine amyloid immunotherapy from this unique standpoint.
Much remains unknown about the precise nature of AD pathogenesis and the role of amyloid immunotherapy. In our opinion, two key issues should be addressed. First, it must be determined whether Aβ remains the critical pathogenic and therapeutic target in AD. Certainly, all recent Aβ immunotherapy AD trials are based on the amyloid cascade hypothesis which places Aβ centrally in AD pathogenesis [9]. Yet, because of failed Aβ immunotherapy casting doubt on traditional hypotheses, interest has shifted toward tau as the next therapeutic target [10]. Beyond Aβ and tau, however, other novel APs might also interact with and be critical to AD pathogenesis. Targeting these may turn out to be essential for linking reduced AD pathology to a clinically meaningful therapeutic outcome. In this regard, our view of amyloidogenic evolvability suggests that a number of APs might be involved in evolvability against multiple brain stressors, and that neurodegenerative disease might be a result of antagonistic pleiotropy derived from amyloidogenic evolvability [8]. In addition to Aβ and tau, several other APs have been characterized in AD brain, including p53 [11], amylin [12], and adrenomedullin [13], suggesting that immunotherapy against a singular AP might be insufficient to generate a clinical benefit.
Second, although the design of amyloid immunotherapy trials has been successively optimized over time, challenges remain. Initially, since the timing of treatment in previous Aβ immunotherapy occurred too late in the disease course to exert any benefits for the neurodegenerative process, later passive Aβ immunotherapies have been staged progressively earlier into mild cognitive impairment and eventually into asymptomatic stages such as in the dominantly inherited AD cohort [14]. While two monoclonal antibodies failed to improve cognition in phase II/III trials, one monoclonal antibody, aducanumab, co-developed by Eisai and Biogen, was controversially approved by the US Food and Drug Administration with a restricted use [15]. However, according to our evolvability hypothesis, one may be concerned that increasingly earlier initiation of disease-modifying therapy for AD treatment/prevention may extend into younger reproductive life, negatively affecting amyloidogenic evolvability (Fig. 1B).
Yet, given that amyloid immunotherapy during younger reproductive life might suppress protofibril formation of APs, leading to reduced amyloidogenic evolvability, this could adversely increase susceptibility to stressors in offspring, causing various pathologies. For instance, it is probable that psychiatric disease including schizophrenia might manifest in offspring due to the decreased Aβ evolvability [16]. Furthermore, neuropathic types of lysosomal storage disorders, such as Niemann-Pick disease and Gaucher disease, might be attributed to decreased AP evolvability, involving Aβ and α-synuclein [17]. Notwithstanding the importance of a possible transgenerational effect of amyloid immunotherapy on offspring/youth, no studies have yet addressed this question. This should not only be assessed using in vivo models, but also more importantly in follow up assessments of patients already treated with amyloid immunotherapies and especially their offspring. Similar concerns may also impact therapies against other APs such as tau, affecting their evolvability. Thus, in attempts to improve the effectiveness of Aβ immunotherapies by applying them earlier in life, they might inadvertently interfere with amyloidogenic evolvability and be detrimental to the brains of offspring, prompting a need to re-evaluate such therapeutic strategies.
Then, how can the aggregation of multiple APs be simultaneously inhibited? Certainly, attacking multiple APs using specific immunotherapies is possible, but might prove overly complex. A more practical target might be activin, a TGF-β family member signaling through type II and -I receptors [18], which is involved in growth and differentiation in a variety of biological systems, and where activin is negatively regulated by follistatin. Supposing that activin might be involved in protein aggregation in AD and stimulation of inflammatory mechanisms via serine/threonine phosphorylation [19], such activin activity of promoting neurodegeneration during aging might reflect evolvability in reproduction through antagonistic pleiotropy. As such, down-regulation of activin would be desirable, perhaps through suppression by follistatin.
Furthermore, recent evidence suggests that the “adiponectin paradox” might be involved in the progression of aging-associated chronic diseases, including AD. Despite its beneficial properties, such as sensitizing insulin signaling, stimulating mitochondrial biogenesis, and suppressing inflammation, adiponectin worsens aging-associated disorders, generating the “adiponectin paradox” [20–22]. Since activin is likely positioned downstream from the “adiponectin paradox”, the activin signaling pathway might be a relevant therapeutic target [23]. Activin might also be centrally involved in antagonistic pleiotropy in aging. Specifically, activin signaling blockade using follistatin, the activin-binding protein, or its analogues [24, 25], might be beneficial in AD (Fig. 1C), as well as in other aging-associated chronic disorders such as muscular dystrophy [26]. In conclusion, taking into account the physiological properties of APs, including potentially antagonistic pleiotropy and relevant interactions, will be critical when designing any AD therapy strategy, and mechanisms that address multiple APs such as activin signaling, might prove therapeutically significant.
Footnotes
ACKNOWLEDGMENTS
We are grateful for the continuous encouragement of Drs. Kaori Hashimoto (Tokyo Metropolitan Institute of Medical Science) and Maria del Carmen Ruiz de la Cruz (University of Chicago).
FUNDING
The authors have no funding to report.
CONFLICTS OF INTEREST
Gilbert Ho declares that he has previously served on an advisory board for Biogen. The remaining authors declare no conflicts of interest.