
Abstract
Despite the apparent neurotoxicity of amyloid-β (Aβ), recent clinical trials of Aβ immunotherapy have not shown any clinical benefit in Alzheimer’s disease (AD). Given this, clarification of the next generation therapeutic strategy in AD is warranted. Hypothetically, adiponectin might be involved in promoting amyloidogenic evolvability in reproduction, which may result in the adiponectin paradox through antagonistic pleiotropy mechanism in aging, leading to AD. Accordingly, preventing the adiponectin paradox by suppressing adiponectin signaling might prove therapeutic in AD.
Keywords
Although the amyloid cascade hypothesis (ACH) has placed amyloid-β (Aβ) at the center of Alzheimer’s disease (AD) pathogenesis, recent clinical trials of Aβ immunotherapy in AD have been unsuccessful, casting doubt on the ACH [1]. Initial human trial of Aβ immunotherapy used active immunization of recombinant Aβ42, but the trial was suspended due to side effects such as encephalomyelitis [2]. Although histological analysis of autopsy brains from Aβ immunized patients were free from Aβ pathology [3, 4], no symptomatic improvement in dementia was observed. Subsequently, several passive Aβ immunotherapies using either monoclonal or polyclonal Aβ anti bodies were tested in humans, also without success [5]. As a group, Aβ immunotherapy has largely not demonstrated efficacy, and one possible rationale for such failure was that timing of the treatment intervention was too late in the disease course. Accordingly, passive Aβ immunotherapies were initiated from the asymptomatic early stage of dominantly inherited AD [6]. An exception, aducanumab, resulting from a collaborative effort between Eisai and Biogen, although controversial, is still under consideration by the US Food and Drug Administration for potential approval [7]. Yet, two other monoclonal antibody therapies, gantenerumab and solanezumab, developed by Roche and Eli Lilly respectively, had failed to improve cognition in Phase II/III trials [8, 9].
Based on the dissociation of protein aggregation of Aβ from cognitive deficits [10], it was predicted that tau, another pathological hallmark of AD, might be a candidate as the next therapeutic target of AD [11], since tau has been implicated in the synaptic pathology and cognitive dysfunction [12]. Consistently, it was recently described that specific phosphorylation sites of tau were associated with structural, metabolic, neurodegenerative and clinical markers of AD [13, 14]. Currently, a phase II clinical study of tau immunotherapy in AD is in progress, including both active and passive immunotherapy [15, 16]. The basic idea of tau as a therapeutic target, however, remains similar to the ACH in that both are based on the pathological functions of amyloidogenic proteins (APs).
In addition, a better understanding of the physiological function of Aβ might be necessary for successful therapy development in AD. As such, we recently proposed that evolvability might be a physiological function of APs relevant to neurodegenerative diseases, such as Aβ in AD and α-synuclein in Parkinson’s disease [17, 18]. Evolvability is the ability of a population of organisms to not merely generate genetic diversity, but to generate adaptive genetic diversity, and thereby evolve through natural selection [19]. Namely, evolvability is defined as the capacity of a system for adaptive evolution [19]. More specifically, the diverse β-sheet structures of protofibrillar APs forms that are found within the intrinsically disordered structures might confer resistance against multiple stressors in parental brains. Subsequently, the protofibrillar APs may be transmitted to offspring through germ cells in a prion-like manner [17, 20]. By virtue of the stress information of APs derived from parental brains, an off spring’s brain can better cope with forthcoming stresses to avoid developmental disorders. On the other hand, neurodegeneration may manifest in parental brains through the antagonistic pleiotropy mechanism in aging. Hereafter, we refer to our view as the evolvability hypothesis (EVH). If EVH is validated, it is expected that EVH might provide clues for AD therapy.
Since recent evidence suggests that the adiponectin paradox might be involved in the pathogenesis of AD [20], we speculated that this phenomenon might be relevant to evolvability. Adiponectin is a cytokine produced by adipocytes that is involved in regulation of energy balance and metabolism as well as other multiple functions, including sensitization of insulin receptor signaling, stimulation of mitochondria biogenesis, and suppression of inflammation [21]. Not withstanding the protective property of adiponectin in many experimental systems, adiponectin is detrimental in aging-associated circulatory diseases, including chronic heart failure and chronic kidney disease, the so-called APN paradox [20]. In the nervous system, adiponectinis protective against neurotoxicity caused by MPP+ (1-methyl-4-phenyl-pyridinium) and Aβ in vitro, and kainic acid-induced excitotoxicity in vivo [21]. Consistent with these findings, adiponectin has been shown to be anti-neurodegenerative in mouse models. For instance, adiponectin was protective against α-synuclein-induced neurodegeneration [22]. Furthermore, osmosin, a plant homologue of adiponectin, was protective against Aβ toxicity [23]. Moreover, knockout of a gene for adiponectin receptor in mice resulted in neurodegeneration [24].
Despite these beneficial properties of adiponectin, adiponectin might also be detrimental in the brain. Supporting this, a recent prospective cohort study in elderly showed that serum adiponectin levels were associated with the severity of amyloid deposits and cognitive deficits, suggesting that adiponectin might stimulate formation of Aβ amyloidosis in the elderly [25]. Moreover, serum adiponectin levels were associated with worse cognitive function in postmenopausal women, which was recently confirmed in another study [26, 27]. On the other hand, increased levels of serum adiponectin were independently reported in AD [21]. Meanwhile, histopathological studies of the postmortem AD brains revealed that adiponectin was sequestered by phospho-tau into the neurofibrillary tangle, implying that tau aggregation was stimulated by adiponectin [28]. Collectively, adiponectin is neuroprotective and anti-neurodegenerative in reproduction, but may promote AD in aging, otherwise known as the adiponectin paradox in AD [20]. We predict that the adiponectin paradox in AD might be derived from the stimulatory effect of adiponectin on amyloidogenic evolvability in reproduction through antagonistic pleiotropy mechanism (Fig. 1). Given that the effect of adiponectin on evolvability in reproduction is beneficial for offspring, adiponectin paradox, the detrimental phenomenon in aging, persists against the pressure of natural selection in evolution.
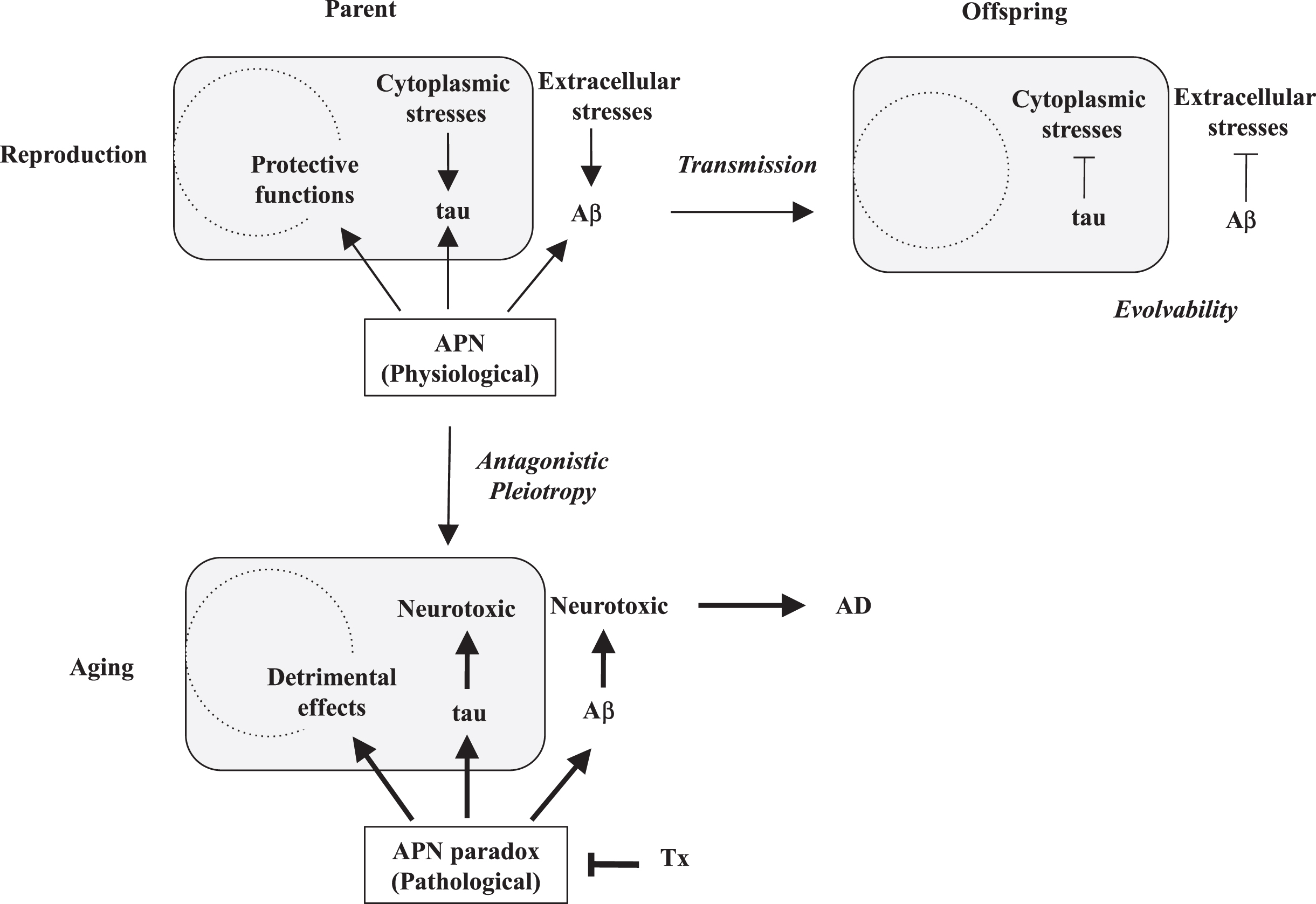
Schematic of the APN regulation of amyloiogenic evolvability and AD. According to EVH, Aβ is involved in evolvability against stressors in the extracellular spaces, while tau is against the stressors in the cytoplasm in parental brain. In response to multiple stressors, aggregation of APs, including Aβ and tau, is induced, and the resulting protofibrils of APs might confer the stress resistance. Subsequently, the APs protofibrils might be transmitted to off spring via germ cells. By virtue of the information carried by the transmission of APs protofibrils in reproduction, off spring can cope with the forth-coming stresses in the brain to escape from neurodevelopmental diseases. Thus, the APs protofibrils might be involved in evolvability against stressors in brain, which is evolutionally beneficial. However, the evolvability of APs protofibrils may increase the risk of AD through the antagonistic pleiotropy in aging. APN may be neuroprotective and stimulate evolvability by the APs protofibrils in reproduction. However, APN might stimulate the development of AD in aging, namely APN paradox. Therefore, decrease of APN expression could be therapeutic (Tx).
At present, the precise mechanism of the adiponectin paradox in AD remains obscure. Because aging-associated circulatory diseases, such as chronic heart failure and chronic kidney disease, are associated with adiponectin paradox [20], in which adiponectin resistance may be multi factorial, integrating abnormalities emanating from insulin signaling, mitochondrial biogenesis, and ceramide metabolism [29], a similar mechanism might be applied to the adiponectin paradox in AD. We predict, however, that adiponectin resistance in evolvability might be a physiological phenomenon, which is distinct from adiponectin resistance in the pathogenesis of AD. Notably, insulin resistance is physiologically important to protect cardiovascular tissues from nutrient-induced injury [30]. Thus, both adiponectin- and insulin resistance might be able to act as adaptive defense mechanisms. Increased levels of adiponectin due to adiponectin resistance may result in more frequent interaction with APs, leading to neurodegeneration in AD. Further supporting the notion of the role for adiponectin in promoting AD, it was established that mid-life obesity is an established risk factor for AD, whereas late-life obesity has been proposed as a protective state [31]. Since expression of adiponectin is down regulated in obesity, the adiponectin paradox and the obesity paradox in AD risk might appear contradictory, but could in fact, represent the same phenomenon.
Given that adiponectin increases protein aggregation and cognitive deficits in the adiponectin paradox, leading to AD, it is reasonable to predict that the adiponectin signaling might be a therapeutic target (Fig. 1). At the ligand level, dose-reduction of adiponectin might be considered. For this purpose, antisense oligonucleotide strategy could be used to reduce the adiponectin mRNA [32], or immunotherapy could suppress adiponectin protein expression. At the receptor level, antagonist-mediated inhibition of adiponectin receptor signaling pathway might also be effective. Antagonists could be designed by molecular docking methods as previously described [33].
Although adiponectin might be promising as a next generation target for AD therapy, there are many issues to be unraveled. In particular, it should be recognized that the effects of adiponectin depend on the stage of AD. In early stages, adiponectin might be neuroprotective with multiple beneficial functions, while adiponectin might be detrimental, promoting the adiponectin paradox in later disease stages. Therefore, it is important to examine whether decreased adiponectin might turn out to be a “trade off.” Conversely, adiponectin might be used as a biomarker to assess the stage of AD. Thus, further investigations are warranted for these issues.
In summary, adiponectin might serve as a driving force of AD, and suppression of the adiponectin paradox might be therapeutic for AD. Thus, a better understanding of the role of adiponectin in evolvability in reproduction and neurodegeneration in aging may pave a new avenue for the therapy of AD.