
Abstract
Iron accumulates in the brain of subjects with Alzheimer’s disease (AD). Here it promotes the aggregation of amyloid-β plaques in which it is abundant. Iron induces amyloid-β neurotoxicity by damaging free radicals and causing oxidative stress in brain areas with neurodegeneration. It can also bind to tau in AD and enhance the toxicity of tau through co-localization with neurofibrillary tangles and induce accumulation of these tangles. Porphyromonas gingivalis is a key oral pathogen in the widespread biofilm-induced disease “chronic” periodontitis, and recently, has been suggested to have an important role in the pathogenesis of AD. P. gingivalis has an obligate requirement for iron. The current paper suggests that P. gingivalis seeks the AD brain, where it has been identified, to satisfy this need. If this is correct, iron chelators binding iron could have beneficial effects in the treatment of AD. Indeed, studies from both animal AD models and humans with AD have indicated that iron chelators, e.g., lactoferrin, can have such effects. Lactoferrin can also inhibit P. gingivalis growth and proteinases and its ability to form biofilm.
INTRODUCTION
Iron can be found in the human brain where its levels increase with age [1, 2]. Importantly, iron acc-elerates the aggregation of the amyloid-β (Aβ) peptide [3–6] and is abundant in Aβ plaques [7]. This may enhance the risk of Alzheimer’s disease (AD) where Aβ plaques are one of the hallmarks [8]. Ayton et al. [9] were the first to demonstrate a relationship between iron and Aβ deposition in a longitudinal clinical study on AD. According to Chung et al. [10], iron, after binding to Aβ, regulates Aβ toxicity in the central nervous system (CNS). Iron induces Aβ neurotoxicity by producing free radical damage and oxidative stress in brain areas with neurodegeneration, probably by promoting aggregation of Aβ [11, 12]. Accordingly, iron can induce Aβ aggregation and has been found to be directly associated with Aβ plaque in vitro and in vivo [8].
In biology, iron cycles between ferrous (Fe2 +) and ferric (Fe3 +) states. This chemistry is used by cells for many redox reactions but can cause deleterious oxidative stress which contributes to AD [8]. Specifically, Fe3 + accumulates in Aβ plaques and ne-urofibrillary tangles (NFTs) of AD brains and is the more potent of the two ions in neurotoxicity [2]. Everett et al. [13] found Aβ capable of accumulating Fe3 + within their aggregates, which resulted in Aβ-mediated reduction of Fe3 + to a redox-active iron (2 +) phase. According to these authors, the ability of iron to form redox-active phases from ferric precursors provides an origin both for the redox-active iron seen in AD tissue and the increased levels of oxidative stress characteristic of AD.
Additionally, ferritin, which is the major iron-bi-nding protein of brain cells [11], is associated with deposition of amyloid. Increased neuronal iron even increases the synthesis and processing of the amy-loid-β protein precursor to produce Aβ [12, 15]. Increased brain iron is associated with plaque-ir-on loading and microglial iron inclusions [16]. Sti-mulation of cultured microglia with interferon γ and Aβ made iron accumulate in these cells [17]. The microglia became glycolytic and iron-retentive, and their phagocytic and chemotactic function was redu-ced. In microglia of mice, iron accumulation trigger-ed a cascade of events that caused altered metabolism and compromised function [17]. Dysregulation of iron homeostasis causing accumulation in neurons can promote changes in Aβ and tau (the second hallmark of AD). According to Gibbons et al. [18], unique misfolded conformations of tau, referred to as strains, have been hypothesized to underlie the distinct neuroanatomical and cellular distribution of pathological tau aggregates. These authors detected a specific tau strain in AD by using immunohisto-chemistry with novel conformation-selective tau antibodies that distinguished it from non-AD tauopathies. Aβ and tau can also disrupt iron homeostasis [2]. This suggests a positive-feedback pathogenic process in AD.
It has been suggested that iron can bind to tau protein(s) in AD and affect their toxicity through co-localization with NFTs [19, 20]. Like Aβ pla-ques, iron can induce accumulation of abnormally phosphorylated tau [20, 21]. Iron increases hyperphosphorylation of tau by stimulating cyclin kinase 5 (CDK5) and glycogen synthase kinase (GSK–3β) activity and reduces the activity of phosphatase 1A [8]. The latter is the major tau phosphatase in host cells [22, 23].
DYSREGULATED IRON HOMEOSTASIS AND ALZHEIMER’S DISEASE
AD is the most common form of neurodegenerative diseases. As mentioned, the two neuropathological hallmarks in AD are extracellular aggregations of Aβ peptide and cytoplasmic inclusions within neurons called NFTs. The latter are microtubule-associated proteins composed of filaments of hyperphosphorylated tau. Both types of hallmark proteins are pro-bably acting synergistically in the pathogenesis of AD (for a review, see [2]).
A causative interplay between dysregulation of iron homeostasis and amyloid plaque formation has been suggested [16]. The authors hypothesized that iron dysregulation and Aβ plaque pathology act syn-ergistically in the process of neurodegeneration and can start a downward cascade of events ending up with manifestation of AD. In an APP(swe)/PS1(deltaE9) transgenic mouse model of AD, light micros-copy revealed that onset of plaque formation occurred at 8 months’ age. Detectable traces of free iron, but no ferritin, were seen around plaques at this age, while their accumulation in and around Aβ plaques had increased at 13 months. Ferritin accumulated mainly at the edge of Aβ plaques, while a smaller amount of free iron was observed in the plaque-free tissue, as well as in and around Aβ plaques [24]. Secreted ferritin, which reflects the intracellular iron load, was increased in the cerebrospinal fluid of individuals with mild cognitive impairment and may be used to predict a near-term progression risk of the disease [2]. Accordingly, free iron and ferritin accumulation seem to follow the formation of amyloid plaques. Another study showed that iron homeostasis is changed in AD patients with lower iron in their serum and an iron overload in several specific brain regions [25].
Iron can induce abnormalities in signal transduction processes related to oxidative damage [26]. Dysregulation of the brain iron homeostasis results in the production of reactive oxygen species, generating reactive aldehydes, which, together with further oxidative insults, cause oxidative modification of proteins, manifested by carbonyl formation [27]. The-se misfolded and damaged proteins overwhelm the ubiquitin/proteasome system, leading to accumulation of the characteristic inclusion bodies seen in many neurodegenerative diseases [27].
AD is characterized by dementia resulting from selective loss of neurons in the hippocampus and cortex [2]. Hare et al. [28] reported increased intrusion of iron into the gray matter of the AD brain compared to controls. This indicated either loss of iron homeostasis in this vulnerable brain region or increased inflammatory processes as a response to chronic neurodegeneration. The authors also observed a trend to increased iron within the white matter of the frontal cortex, potentially due to disrupted iron metabolism preceding loss of myelin integrity.
Accordingly, an imbalance in brain iron has been established in AD. This imbalance can even affect expression of and response to inflammatory agents [29] like microorganisms. Deregulation of iron homeostasis also causes generation of reactive oxygen species that can underlie an increased oxidative stress burden [15] or lead to oxidative damage [19]. Similar to accumulation of iron, oxidative stress is an early event in the pathogenesis of AD.
The source of the excess iron in the AD brain is unclear. However, over the last years, neuroimaging studies have increasingly implicated micro- and macro-structural abnormalities in the white matter in the risk and progression of AD, suggesting that in addition to the neuronal pathology characteristic of the disease, white matter degeneration and dem-yelination can be important pathophysiological characteristics. One theory involves oligodendrocytes which have the highest iron content of all cell types. Their iron content increases with age and even further in AD [30]. The high iron content and a low antioxidant content make oligodendrocytes most vulnerable to oxidative stress in the CNS. If oxidative stress is increased by age, or AD, it may lead to increased cell damage [31, 32].
Another possible source of excess brain iron is the black pigment within the outer membrane of P. gingivalis which carries bound [Fe(III)PPIX]2O on its surface [33]. On invasion into the CNS by P. gingivalis these Fe-containing compounds will follow and exert toxicity towards the host’s brain cells. The cell surface muco-oxy dimer layer formed may also serve as a protective barrier against reactive oxidants and permit P. gingivalis to maintain a local anaerobic environment.
IRON AND SENESCENCE
AD is prevalent in elderly people due to the biological aging process. Iron dysmetabolism is closely connected to cellular senescence and loss of aerobic glycolysis (for a review, see [34]). Iron induces DNA damage and endothelial cell senescence that increase the permeability of the blood-brain barrier and the risk of microbial translocation to the brain [34–36]. Moreover, iron activates NLRP3 inflammasomes, linking this biometal to AD inflammation [37], dysfunctional mitochondria, and impaired mitophagy [38–40]. The level of the iron storage protein, ferritin, is thought to be a reliable senescence marker, yielding support to the concept of ferrosenescence [41]. This has given credit to a new, unproved hypothesis, suggesting that age-related increase in the free iron pool resuscitates dormant microbes in the brain parenchyma [42].
PORPHYROMONAS GINGIVALIS AND ALZHEIMER’S DISEASE
Recent research has opened the possibility that AD can be caused by infection and neuroinflammation accompanying the former. A field receiving increasing attention recently has been “chronic” periodontitis which is a common oral disease in the adult population all over the world. Periodontal pathogens and their main virulence factors like lipopolysaccharide (LPS) and gingipains have been detected in the brain of AD patients and animal models of AD [43–48]. Dominy et al. [44] found P. gingiva-lis DNA and gingipain antigens in human AD brains and showed in mice that oral administration of small-molecule gingipain inhibitors blocked gingipain-induced neurodegeneration. The findings in the human brains appeared even before AD manifested and progressed further and could therefore not be a result of inadequate oral hygiene accompanying AD. P. gingivalis could also be detected in the cerebrospinal fluid of AD patients confirming that the CNS had been affected. These authors [44] also found that RgpB localized with neurons, astrocytes, and pathology in the hippocampus, and Kgp in the cerebral cortex of AD patients. So far, this is the strongest indication that AD can be associated with “chronic” periodontitis where P. gingivalis is a keystone bacterium [49–51].
PORPHYROMONAS GINGIVALIS AND IMMUNITY
Virtually all human pathogens have an absolute requirement for iron. An important task of innate immunity is to limit iron availability to invading microorganisms (nutritional immunity). To be successful in causing disease, human pathogens must have mechanisms to circumvent nutritional immunity [52]. An important pathogen in the oral microbiota is P. gingivalis which in addition to being a keystone pathogen in periodontitis [49–51], has recently been associated with AD [43, 53–56]. This bacterium is a master of immune subversion [57, 58], probably also in AD [59]. Central in this task is its strategy to overcome iron-withholding defenses. Of note is also that immunity can be declined in AD due immunosenescence [60].
PORPHYROMONAS GINGIVALIS AND ITS REQUIREMENTS FOR IRON
P. gingivalis, which is a heme-dependent bacte-rium, lacks siderophores. Instead, it uses specific outer membrane receptors, proteases (particularly gi-ngipains), and lipoproteins to acquire iron/heme from the environment [61, 62]. P. gingivalis can even use heme acquisition systems of accompanying bacteria to satisfy its own requirements [63].
Specific proteins are operating in P. gingivalis for iron/heme capture [60] and specific genes for iron/heme utilization [64]. Outer membrane vesicles, containing gingipains, are crucial for both micro- and macro-nutrient capture of iron for P. gingivalis, especially of heme [65].
P. gingivalis has an enzyme system which opens the heme ring for iron release. Thus, Daspher et al. [66] characterized a novel outer membrane hemin-binding protein in P. gingivalis: IhtB (iron heme transport, formerly designated Pga30). The authors found this protein, released from the cell surface, to bind hemin by using hemin-agarose. Analysis of the deduced amino acid sequence of IhtB showed significant similarity to the Salmonella typhimurium protein CbiK which is a cobalt chelatase structurally related to the ATP-dependent family of ferrochelatases. The authors suggested that IhtB is a peripheral outer membrane chelatase that could remove iron from heme prior to uptake by P. gingivalis. This hypothesis needs confirmation. Since it is not clear if iron removed by IhtB actually is taken up by the cell, this could potentially add to the free iron pool of the AD brain. P. gingivalis probably expresses other proteinaceous binding sites which are important in the binding/transport of heme/hemin from the host, for example A-LPS [67]. Others have assumed that uptake of heme in P. gingivalis may require binding of heme to Kgp and thereafter binding of Kgp to the heme/hemoglobin receptor HmuR [68].
Goulet et al. [69] reported that P. gingivalis gingipains also degrade human transferrin, providing sources of iron and transferrin peptides. The iron-containing fragments or the release of iron itself was suggested to contribute to tissue destruction by catalyzing the formation of toxic HO*.
P. gingivalis also uses hemoglobin as an iron source. Notably, this source is used much more effectively than other iron-containing compounds under an iron-limited environment [70].
PORPHYROMONAS GINGIVALIS MAY SEEK THE BRAIN TO ACQUIRE HEME
Heme is necessary for P. gingivalis to survive in its site of origin (the periodontal pocket) and probably also in its peripheral site (the AD brain). The main sources of heme for P. gingivalis in the mouth are hemoproteins in the saliva and gingival crevicular fluid and erythrocytes. In the AD brain, heme is probably bound to Aβ in a complex. Recently, it was found that the formation and enzymatic activity of Aβ-heme complexes are pathological key features of AD [71, 72] (Fig. 1). Accordingly, free heme and amyloid-β could be a dangerous liaison in AD.
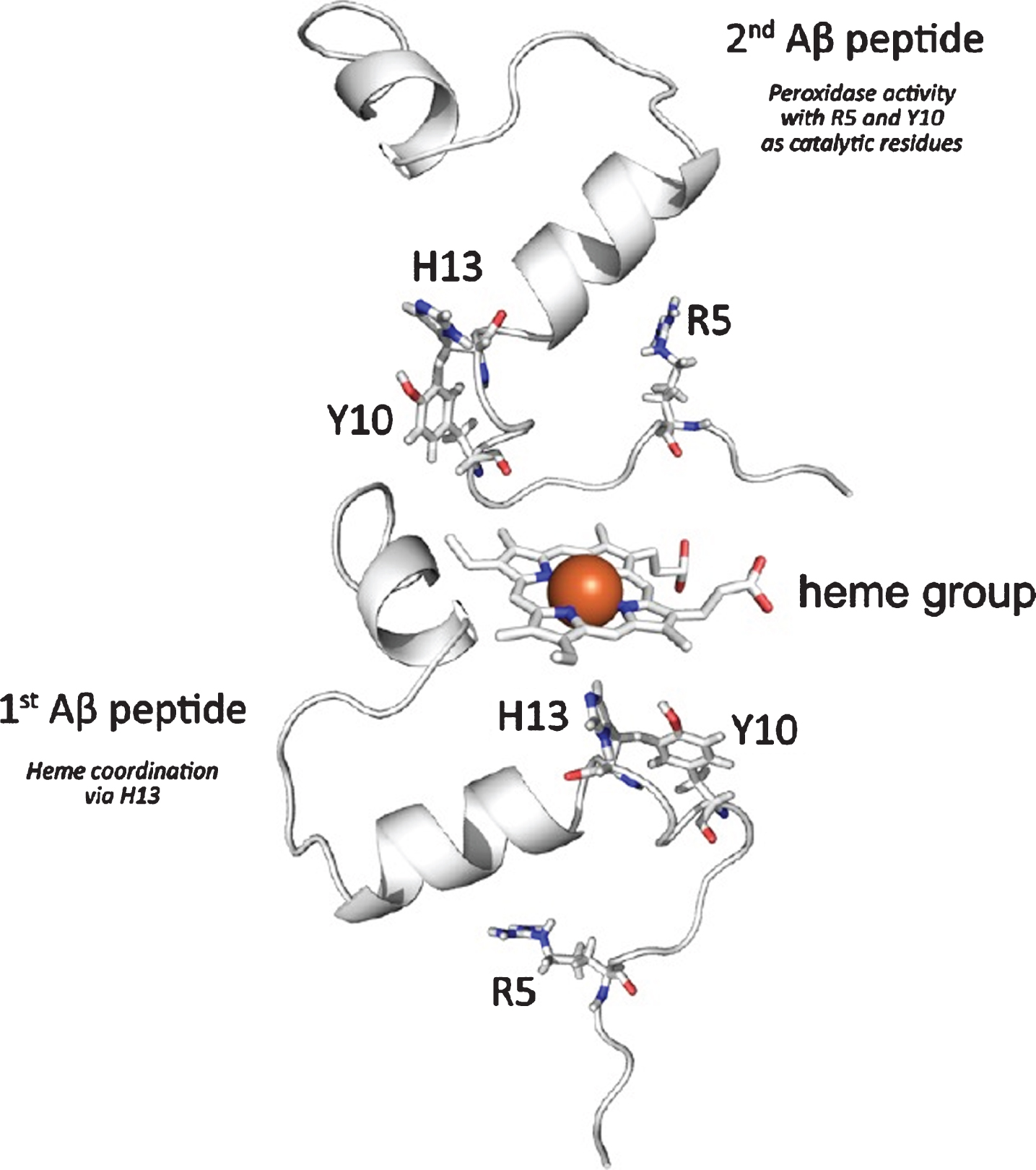
Schematic presentation of amyloid β (Aβ)-heme complex formation based on published investigations on the complexation of free heme by Aβ. Interaction is proposed to mainly take place between heme and the N-terminal part of the peptide. While one peptide is mainly involved in the coordination of the heme iron at the distal heme site via H13, a second peptide may coordinate at the proximal heme site to provide R5 and Y10, which are responsible for the subsequent peroxidase activity of the complex. Collected from [72].
By lysing Aβ plaques, which in AD are overloaded with iron, P. gingivalis probably achieves access to iron either as heme, free iron or ferritin (one of the intracellular iron-storage proteins of P. gingivalis). Ferritin accumulates mainly at the edge of Aβ plaques, while a smaller amount of free iron is observed in the plaque-free tissue, as well as in and around Aβ plaques [24]. Whether P. gingivalis (1) lyses plaques through their gingipains, (2) acquires iron in the plaque surroundings, (3) extracts iron from plaque-free brain tissue, or (4) takes it from ferritin or microglia, the consequences of acquisition of iron from these sources may aggravate the iron dyshomeostasis existing in the AD brain. Aberrations in the brain iron homeostasis can increase the levels of this redox-active metal, causing dislocalization of the metal and catastrophic oxidative damage to sensitive cellular and subcellular structures [73]. Iron levels are lower in serum of AD patients than in Aβ plaques [25]. Aβ plaques may therefore be a preferred peripheral site of P. gingivalis for iron supplementation rather than serum.
CONCLUDING REMARKS
It seems plausible that one reason for P. gingivalis seeking the brain and establishing itself here is its requirements for iron. This could occur in the brains of healthy old people or at the mild cognitive impairment stage, or later in the AD process. As suggested previously, a 10-year lag phase of “chronic” periodontitis is needed for this widespread oral disease to become a risk factor for AD [55]. However, P. gingivalis may possibly also contribute to the free iron pool in the AD brain through its bound [Fe(III)PPIX]2O and IhtB. Presence of P. gingivalis in the brain could tip the delicate iron balance here causing iron dyshomeostasis and also promote development of AD through its shed inflammagens such as LPS and proteolytic enzymes transferred through the blood brain barrier by its outer membrane vesicles [74]. This imbalance may affect expression of and response to inflammatory agents [29] such as brain-residing microorganisms. Age-related increase in the free iron pool may also resuscitate dormant microbes in the brain parenchyma which could be another important feature of AD [42]. The growth of these organisms in vivo is limited by a lack of free iron, and iron dysregulation could be an important factor in their resuscitation [75–77]. Iron dysregulation and microbial aberrations could also affect the hematological system by promoting fibrin amyloidogenesis and pathological clotting, paving the way for iron-induced clotting in the development of AD [78].
As a consequence of the high levels of iron in AD, iron chelators have been tested in the treatment of AD in rodents and man [15, 80]. Iron chelators caused reduction in Fe3 +-induced Aβ42 accumulation in vitro [81], in tau aggregates from AD patients, and in slices of human AD brains [82, 83]. In genetic mouse models of AD, iron chelators reduced cognitive impairment together with GSK3β activity, tau phosphorylation, Aβ generation and accumulation of Aβ in the hippocampus. Iron chelators in AD treatment of other rodent models have also provided encouraging results (reviewed by D’Mello and Kindy [2]). Even oxidative stress and activation of microglia have been diminished by iron chelators. Of particular note, sustained administration of iron chelators to patients with early AD slowed the progression of dementia [84].
Molecules of special interest as iron chelators are lactoferrin (LF) and ergothioneine (for reviews, see [85, 86]). These nutraceuticals can act as iron-mopping agents. LF has been found important in the treatment of AD where it may prevent iron deposition and block Aβ-aggregation, tauopathy and neuronal damage [87]. LF can also inhibit P. gingivalis growth and proteinases (Fig. 2) and its ability to form biofilm [88, 89].
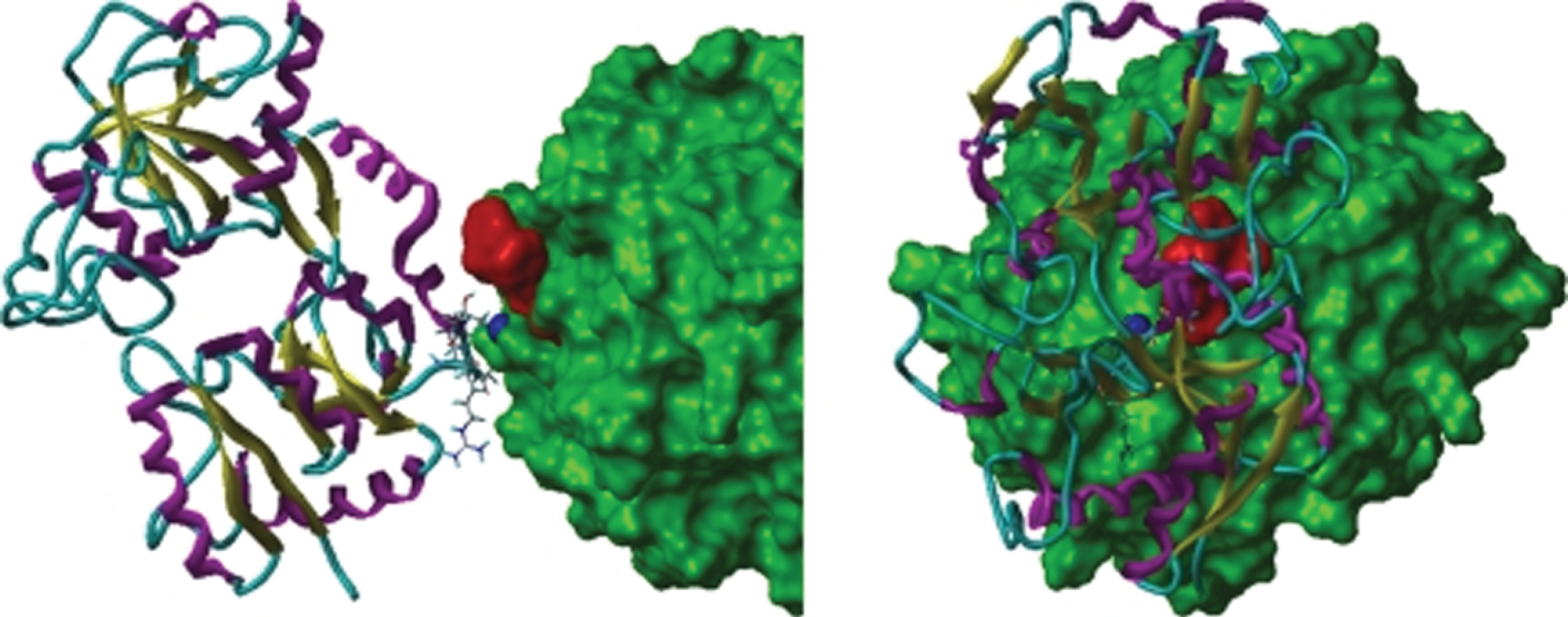
Orthographic view of the modeled lactoferrin (LF)-RgpB complex. The solvent-accessible surface of RgpB is green, the dark blue space-filling atom is the zink ion bound to the catalytic histidine (His244) of RgpB. LF is illustrated as a ribbon structure (β-strand in yellow, α-helix in magenta, and coil in cyan). The side-chains of residues that moved to within 3 Å of RgpB during the dynamics simulation are shown as “capped sticks”. The location of the atoms of the RgpB inhibitor, DFFR-chloromethylketone, is shown by red-space filling atoms (from: [89] Permission granted by RightsLink/ASM)
Iron in the brain seems to be an underappreciated driver of AD disease progression. Its cooperation with bacteria, in this case P. gingivalis, could increase the toxic potential of both in the AD pathogenesis. It seems plausible that iron chelators by binding iron in the AD brain may lessen the signs of AD and simultaneously make the brain less favorable for survival of P. gingivalis and other putative AD pathogens. This hypothesis, previously overlooked, should be confirmed before firm conclusions can be made.
CONFLICT OF INTEREST
The author has no conflict of interest to report.