
Abstract
Diverse pathogens are detected in Alzheimer’s disease (AD) brains. A bioinformatics survey showed that AD genome-wide association study (GWAS) genes (localized in bone marrow, immune locations and microglia) relate to multiple host/pathogen interactomes (Candida albicans, Cryptococcus neoformans, Bornavirus, Borrelia burgdorferri, cytomegalovirus, Ebola virus, HSV-1, HERV-W, HIV-1, Epstein-Barr, hepatitis C, influenza, Chlamydia pneumoniae, Porphyrymonas gingivalis, Helicobacter pylori, Toxoplasma gondii, Trypanosoma cruzi). These interactomes also relate to the AD hippocampal transcriptome and to plaque or tangle proteins. Upregulated AD hippocampal genes match those upregulated by multiple bacteria, viruses, fungi, or protozoa in immunocompetent cells. AD genes are enriched in GWAS datasets reflecting pathogen diversity, suggesting selection for pathogen resistance, as supported by the old age of AD patients, implying resistance to earlier infections. APOE4 is concentrated in regions of high parasitic burden and protects against childhood tropical infections and hepatitis C. Immune/inflammatory gain of function applies to APOE4, CR1, and TREM2 variants. AD genes are also expressed in the blood-brain barrier (BBB), which is disrupted by AD risk factors (age, alcohol, aluminum, concussion, cerebral hypoperfusion, diabetes, homocysteine, hypercholesterolemia, hypertension, obesity, pesticides, pollution, physical inactivity, sleep disruption, smoking) and by pathogens, directly or via olfactory routes to basal-forebrain BBB control centers. The BBB benefits from statins, NSAIDs, estrogen, melatonin, memantine, and the Mediterranean diet. Polymicrobial involvement is supported by upregulation of bacterial, viral, and fungal sensors/defenders in the AD brain, blood, or cerebrospinal fluid. AD serum amyloid-β autoantibodies may attenuate its antimicrobial effects favoring microbial survival and cerebral invasion leading to activation of neurodestructive immune/inflammatory processes, which may also be augmented by age-related immunosenescence. AD may thus respond to antibiotic, antifungal, or antiviral therapy.
Keywords
INTRODUCTION
Multiple and diverse pathogens, have been implicated in Alzheimer’s disease (AD) in epidemiological studies. Meta-analysis studies have reported the association of AD with spirochetal (Odds ratio (OR) = 10.61) or Chlamydia pneumoniae (C. pneumoniae) infection (OR = 5.66) [1] and association of herpes simplex (HSV-1) infection with AD, which is augmented by possession of the APOE4 allele (OR = 2.71) [2]. Periodontitis has also been associated with AD (meta-analysis OR = 1.69 for all periodontitis, and 2.98 for severe periodontitis) [3] and is associated with increased amyloid-β (Aβ) load in patients in vivo [4]. Meta-analysis has also reported a significant association between Helicobacter pylori (H. pylori) infection and dementia (OR = 1.71) [5]. Pathological burden (cytomegalovirus, HSV-1, Borrelia burgdorferi (B. burgdorferi), C. pneumoniae, and H. pylori) rather than any individual pathogen has also been associated with AD [6].
The hepatitis C virus has also been associated with dementia [7] and dementia is part of a spectrum of neurological disorders related to the human immunodeficiency virus, HIV-1 (NeuroAIDS) [8]. Amyloid plaques are found in the brains of HIV-1 patients and Aβ deposition also predict neurocognitive disorders in HIV-1 infected APOE4 carriers [9, 10]. The Borna virus has been associated with a rare form of hippocampal degeneration [11] rather than AD, but does produce extensive degeneration of the cortical and hippocampal cholinergic innervation in rats, analogous to the situation in AD [12]. Borna virus infection of cortical and limbic brain areas produces microglial activation in the hippocampus and neocortex. Aβ deposits decreased in the parenchyma but increased in the walls of cerebral vessels [13].
Epidemiological studies are evidence for a link between infection and AD, but do not necessarily imply actual infection of the brain. Many viruses can enter the brain via olfactory neural pathways to the basal forebrain [14], a route also employed by C. pneumoniae [15] and the cerebral entry of pathogens may also be facilitated by disruption of the blood-brain barrier (BBB) in AD, which appears to be an early feature of the disease [16–18]. Cerebral microbleeds and cortical siderosis (an increase in blood-derived iron deposition) are also a feature related to BBB disruption in AD patients [19–21]. Many bacteria depend upon the availability of free iron and such effects may contribute to their successful colonization in AD [22].
The BBB is also damaged by systemic inflammation [23], or by systemically administered bacterial cell wall components, lipopolysaccharide (LPS) [24], or lipoteichoic acid [25] suggesting that repeated peripheral bacterial infections (a feature of the elderly population) could also weaken the BBB. The BBB can also be damaged by viral infection (vaccinia or HSV-1) [26] or by Porphyrymonas gingivalis (P. gingivalis) [27], while other pathogens such as B. burgdorferi [28], C. pneumoniae [29], or Cryptococcus neoformans (C. neoformans) [30] have found other ways to circumvent this barrier. Pathogen infection in the elderly may also be favored by age-related immunosenescence [31]. This decreasesimmunity-related responses but also results in increases in inflammatory reactions termed “inflamm-aging” [31, 32]. The control of the BBB, the routes of pathogen entry and immunosenescence are covered in greater detail in a later section.
Pathogens that have been detected in the AD brain using DNA detection or immunocytochemistry include HSV-1, C. Pneumoniae, B. Burgdorferi, and many other spirochetes [33–35], human herpes virus 6 (HHV-6) [36], the Epstein-Barr virus (EBV) [37], and Propionibacterium acnes [38]. Bacterial LPS, an immunogenic component of the outer coat of gram-negative bacteria, and E coli K99 (a filamentous fimbrial thread that attaches to other bacteria or host cells) were detected immunocytochemically in brain parenchyma and vessels in all AD (24/24) and control brains (18/18). K99 levels were greater in AD compared to control brains and K99 was localized to neuron-like cells in AD but not control brains. LPS levels were also greater in AD compared to control brain and LPS colocalized with Aβ in amyloid plaques and around vessels in AD brains [39].P. gingivalis LPS has also been detected in AD brains postmortem [40].
Fungal/yeast species detected in the AD brain include: Saccharomyces cerevisiae, Malassezia globosa, Malassezia restricta, Penicillium Phoma, Cladosporium, Neosartorya hiratsukae, Sclerotinia borealis, Candida albicans (C. albicans), and several other Candida species [41, 42]. Fungal DNA and proteins have also been detected in AD cerebrospinal fluid (CSF) [43].
Preliminary microbiome studies using unbiased next-generation sequencing have also identified many fungi in AD brains (Alternaria, Botrytis, Candida, Cladosporium, and Malassezia species [44]. A pilot next-generation sequencing study has also identified several bacterial species in both control and AD brains with an overall higher bacterial load in AD, with a relatively higher proportion of Actinobacteria, and notably Propionibacterium acnes [45]. Evidently, further microbiome studies in blood and brain are warranted for all classes of pathogens and independent confirmation across laboratories is important for individual species.
AD is characterized pathologically by amyloid-containing plaques and Aβ has been extensively implicated in the pathogenesis of AD [46]. The involvement of pathogens in AD has been strengthened by the observations that Aβ is an antimicrobial peptide active against bacteria and fungi [47, 48] and the influenza [49] and herpes simplex viruses [50]. Aβ production in the AD brain may thus be a common response to diverse infections [51].
Indeed, many viruses (HSV-1, HSV-2, cytomeglovirus) [33, 53] or bacteria (C. pneumoniae, H. pylori, Salmonella Typhimurium, B. burgdorferi), and other spirochetes or systemically administered E. coli or Porphyromonas gingivalis-derived LPS increase Aβ production in vitro or in vivo [47, 54– 57], as does the protozoan Plasmodium berghei, which causes malaria in mice [58]. Several of these pathogens also evoke tau phosphorylation [33–35].
The diversity of viruses, bacteria, and fungi contributing to AD, Aβ deposition, or dementia suggests a polymicrobial relationship with these conditions. The broad-spectrum antimicrobial effects of Aβ imply that its recruitment for pathogen defense would similarly relate to many species.
Many AD genes relate to the immune system and to inflammatory processes that are primarily designed to react to infection. They are also localized in immune cells, particularly the B cell lymphocyte [59–62]. AD genes are also predominantly expressed in microglial cells, the resident macrophage/immune cells of the brain [63, 64].
Many AD genes encode proteins that physically interact within a defined protein/protein interaction network, and this network has been subject to positive selection pressure. Selection for pathogen resistance was proposed as one possible explanation [65]. Pathogens have shaped human evolution and the development of the immune and pathogen defense mechanisms, endowing descendants of the survivors of dangerous infections with resistance genes [66–68].
There have been few studies attempting to relate specific AD genes to pathogen resistance. The APOE4 allele is geographically concentrated in tropical regions with a high parasite burden [69] and may also contribute to resistance against Giardia and cryptosporidial diarrhoeal infections in young children [70, 71]. The frequency of this allele is high in sub-Saharan Africa and other areas where malaria is endemic, but while plasma samples from APOE4 subjects inhibit the growth of the malaria parasite Plasmodium falciparum [72], APOE alleles had no major protective effects against malaria in Gambian children [73]. APOE2 has been reported as a potential risk factor for early malaria infection in Ghanaian infants [74]. APOE4 possession may also have a protective effect against hepatitis C infection and enhances viral clearance in infected patients [75]. Old age is the principal risk factor in AD, and this too implies survival from the many infectious diseases that have been and in many regions still are among the greatest causes of death in children and adults[76, 77].
Many AD gene variants from genome-wide association studies (GWAS) also converge towards the common effect of increasing Aβ production [78, 79].In the circulation, amyloid-β protein precursor (AβPP) and γ-secretase are highly expressed in the dendritic cells that scout for invading pathogens [80]. In this context, enhanced Aβ production can also be considered as evolutionarily advantageous [81]. This theme is carried through in postmortem studies showing that neuroinflammatory changes are prevalent in vulnerable regions of the AD brain [82, 83]. The proteins found in plaques and tangles also contain many related to the immune system, inflammation and autophagy, all of which play a general role in pathogen defense [84–86] as does Aβ.
Previous studies have shown that the host genes involved in the life cycles of several pathogens implicated in AD relate to AD susceptibility genes [80]. The proteins found in AD plaques and tangles are also enriched in those used by HSV-1 during its life cycle [84] and the HSV-1 or Toxoplasma gondii(T. gondii)/host interactomes are also enriched in AD susceptibility genes [87, 88].
Similar studies have noted significant overlaps between the EBV/host interactome and diseases in which the virus is implicated, including B cell lymphoma [89] or multiple sclerosis [90]. The interactomes of oncogenic viruses also relate to cancer genes [91] suggesting important gene/environment interactions that may condition disease susceptibility.
In this study, the host pathogen interactomes of 17 fungal, bacterial, viral, and protozoan pathogens were analyzed in relation to 78 AD GWAS genes. The anatomical location of the AD genes was also queried against proteomic/genomic datasets from multiple tissues.
In relation to the potential selection of AD genes for pathogen resistance (e.g., the concentration of the APOE4 allele in areas of high parasite burden), the 78 AD genes were also compared against a series of GWAS datasets related to general pathogen or protozoan diversity, viral diversity and the immune response to parasitic worms, across multiple human populations in different geographical locations. Such genes are likely to have been selected for pathogen resistance [92–95].
The host genes of the 17 pathogen interactomes were also compared with the combined up anddown-regulated genes from a transcriptome study of the AD hippocampus, postmortem [83] and to the proteins found in plaques or neurofibrillary tangles. The upregulated genes from this AD hippocampal study were also compared with upregulated genes from numerous infection microarray datasets (viral, bacterial, fungal, and protozoan) housed at the Molecular signatures database [96] or the Gene Expression Omnibus [97].
Different types of pathogens are detected by a large family of pattern recognition receptors, for example toll-like receptors (TLRs) or nucleotide binding oligomerization domain (NOD)-like receptors, c-type lectin receptors, RIG-I-like receptors, and intra-cytosolic DNA or RNA sensors, inter alia. Each relatively selectively reacts to bacterial or fungal components or to viral or other pathogens’ DNA or RNA [98]. The pathogens addressed in this study each interact with subsets of these and other processes, for example antimicrobial peptides and antiviral genes, specifically related to infection. These pathogen-specific aspects were addressed by a survey of expression changes in the AD blood, CSF or brain, which were matched to the known effects of bacterial, viral and fungal representatives.
Finally, the presence of pathogens in the AD brain may be related to the BBB deficits observed in aging and in AD and to the ability of certain pathogens to circumvent this barrier via olfactory neural routes, or by other means. These aspects are reviewed in relation to the ability of pathogens and other AD environmental risk factors to disrupt the BBB.
The results show that the hippocampal transcriptome genes and the proteins found in plaques and tangles in AD relate to the life cycles and effects of these diverse pathogens and that many AD susceptibility genes also relate to pathogens, but more likely to pathogen resistance than susceptibility. AD genes are also concentrated in immune-related locations and in the BBB. This barrier is defective in AD and it is deleteriously affected by pathogens and by many other non-pathogen environmental risk factors in AD.
MATERIALS AND METHODS
Seventy-eight genes associated with AD (reported genes) were obtained from the NHGRI-EBI Catalog of published GWAS [99], available at: http://www.ebi.ac.uk/gwas, accessed January 2016, version 1.0, using studies labelled as “Alzheimer’s disease” or “Alzheimer’s disease late-onset”. These genes and their relationships with pathogens or the immune system are catalogued in Supplementary Table 1. The genes are highlighted in bold throughout the text.
The tissue and cellular distribution of the 78 AD genes were analyzed using the functional enrichment analysis tool (FUNRICH) [100] (http://funrich.org/index.html) which derives proteomic and genomic distribution data from >1.5 million annotations. It provides the total number of genes in datasets from each region sampled and returns the significance of any enrichment for the uploaded AD genes, using the hypergeometric probability test, with p values corrected using the Storey and Tibshirani method (Q values) [100]. AD gene enrichment was also analyzed in a BBB proteome dataset of mouse cerebral arteries (6620 proteins) [101]. The presence of the AD genes in exosomes, a means of transit through cells allowing intercellular communication [102, 103], was assessed using ExoCarta (http://www.exocarta.org) a manually curated database of exosomal proteins, RNA and lipids [104]. The exosomal pathway is hijacked by several viruses, contributing to intercellular spread and immune evasion [105, 106].
Genes related to general pathogen diversity, protozoan and viral diversity and to the immune response to parasitic worms are from a series of papers concerning evolutionary selection pressure relevant to pathogen resistance [92–95].
The host/pathogen interactomes of two fungal species (C. albicans, C. neoformans), eight viral species (Borna virus, human cytomegalovirus (HCMV), Ebola virus, HSV-1, human endogenous retroviruses HERV-W, HIV-1 (the latter from the HIV-1, human interaction database [107] http://www.ncbi.nlm.nih.gov/genome/viruses/retroviruses/hiv-1/interactions, EBV, hepatitis C and influenza A viruses) four bacterial species (B. burgdorferi, C. pneumoniae, Porphyromonas gingivalis (P. gingivalis), H. pylori) and two protozoans [T. gondii and Trypanosoma cruzi (T. cruzi)] were obtained by literature survey and from existing databases. These referenced interactomes can be accessed at http://www.polygenicpathways.co.uk/HPI.htm.
Genes misregulated in the AD hippocampus are those reported from a postmortem microarray study [83]. Up- and downregulated genes (n = 2,879) were combined for comparison with the pathogen interactomes. The upregulated genes (n = 1,690) from this AD hippocampal study contain the pathways relevant to pathogens and immune activation (inflammation, complement activation and the defense response) [83] and these were chosen for comparison with upregulated genes from infection datasets at the Molecular signatures database (MSigDB; http://software.broadinstitute.org/gsea/msigdb/index.jsp). MSigDB contains several thousand microarray datasets which can be compared against the various AD inputs [96]. Infection related datasets, and those related to TLR ligands, were identified using search terms (e.g., infection, virus, bacteria, TLR, LPS, etc.). Microarray viral infection datasets (upregulated gene sets) from the gene expression omnibus (GEO) [108] were also downloaded from the Harmonizome database (http://amp.pharm.mssm.edu/Harmonizome/) from the Ma’ayan laboratory of computational systems [109]. For the searched gene sets, most of the data outputs were restricted at source (by MSigDb or GEO) to the top upregulated genes (usually ∼ 200– 300).
In addition to this AD hippocampal microarray study, a literature survey was undertaken to analyze the gene or protein expression profiles of components more specifically related to pathogen defense in the AD brain, blood, or CSF. These included TLR- and NOD-like receptors, c-type lectins, antimicrobial peptides, defensins, interferons and viral DNA and RNA detectors, and antiviral oxysterols, inter alia. Many of these are endowed with relative specificity against viruses, bacteria or fungi and where possible their known effects were matched to the pathogens used in this study. These properties are reviewed in Supplementary Table 2.
The proteins found in plaques or neurofibrillary tangles are from two proteomics studies yielding 488 proteins in plaques [85] and 90 in tangles [86].
Assuming a human genome of 26846 coding genes and an interactome or other gene set of N genes one would expect N/26846 to exist in the comparator dataset. For example, when comparing 2879 misregulated AD hippocampal genes against any pathogen interactome one would expect 2879/26846 (10.7% ) to figure in the pathogen interactome. This calculation was used to define expected values and the enrichment values (observed/expected) in relation to other datasets. Significance of the enrichment was calculated using the hypergeometric probability test. The resultant p values from each analysis series were corrected for false discovery (FDR) [110]. Nominally significant FDR corrected values are considered at p < 0.05 and a corrected Bonferroni p value threshold is illustrated on each set of graphs. (Bonferroni P = 0.05/N, where N is the maximum number of possible comparisons for each situation (e.g., 78 AD genes or 1690 upregulated genes in the ADhippocampus).
RESULTS
Immune-related properties of the AD GWAS genes
The properties of the 78 AD genes, specifically in relation to pathogens, or to the immune/inflammatory system are reviewed and referenced in Supplementary Table 1. A number of these genes are primarily concerned with immune function (HLA-DRB1, HLA-DRB5, HMHA1, IGH) while many others with diverse primary effects also possess relevant properties in relation to the immune system (ACE, ADAMTS20, AP2A2, BCL3, BIN1, CR1, CLU, CUGBP2, EPHA1, GAB2, INPP5D, MEF2C, MS4A3, MS4A4A, RIN3, SCIMP, SPPL2A, STK24, TREM2, TREML2, ZNF224) or pathogen defense, e.g., interferon signaling (CD2AP, CELF1, MEF2C, MMP12, PTK2B, TREML2, ZNF224), TLR signaling (DISC1, PTK2B, SASH1, SCIMP) phagocytosis (ABCA7, BIN1, CR1, INPP5D, TREM2), or autophagy (SQSTM1). A number of the AD genes also act as primary receptors for pathogens. These include the poliovirus receptor PVR, the HSV-1 receptor PVRL2, and complement receptor CR1. CD33 binds to the HIV-1 gp120 protein and to diverse forms of sialic acid which coats many pathogens. Others bind bacterial LPS (APOC1, TREM2) or the Escherichia coli cytotoxic necrotizing factor 1 (BCAM). CEACAM16 is a member of a large family of adhesion molecules that bind bacteria or C. albicans, although this particular form is poorly characterized. Others (AP2A2, BIN1, CD2AP, MS4A4A, PICALM, and SORL1) are involved in endocytosis, an obligate requirement for pathogen entry following binding to cognate receptors (see Supplementary Table 1 for references).
AD GWAS genes are enriched in immune locations (Fig. 1)
The AD genes are most significantly enriched in the exosome and bone marrow datasets. As noted above, exosomes are hijacked by many viruses for intercellular spread. Exosomes are prevalent in plasma [111] (also enriched in AD genes) and are also the means by which intracellularly generated Aβ is conveyed to the extracellular space [112]. The bone marrow is the hematopoietic source of red and white blood cells and platelets [113], and bone marrow B cells rapidly respond to infection. The parathyroid gland expresses many AD genes and also plays a role in hematopoiesis [114, 115]. Other immune related areas enriched in AD genes include the appendix, spleen, tonsils, the lymph nodes, and the bronchus and neutrophils. The appendix is an important component of mucosal immune function, particularly B cell-mediated immune responses and extrathymically derived T-lymphocytes [116]. The tonsils and nasopharynx, also enriched in AD genes, play an important role in the initial defense against respiratory pathogens [117] and against those capable of entering the brain via the olfactory route. These include Herpes viruses (HSV-1 and HHV6), the Borna disease virus, the influenza A virus [14], and C. pneumoniae [118].
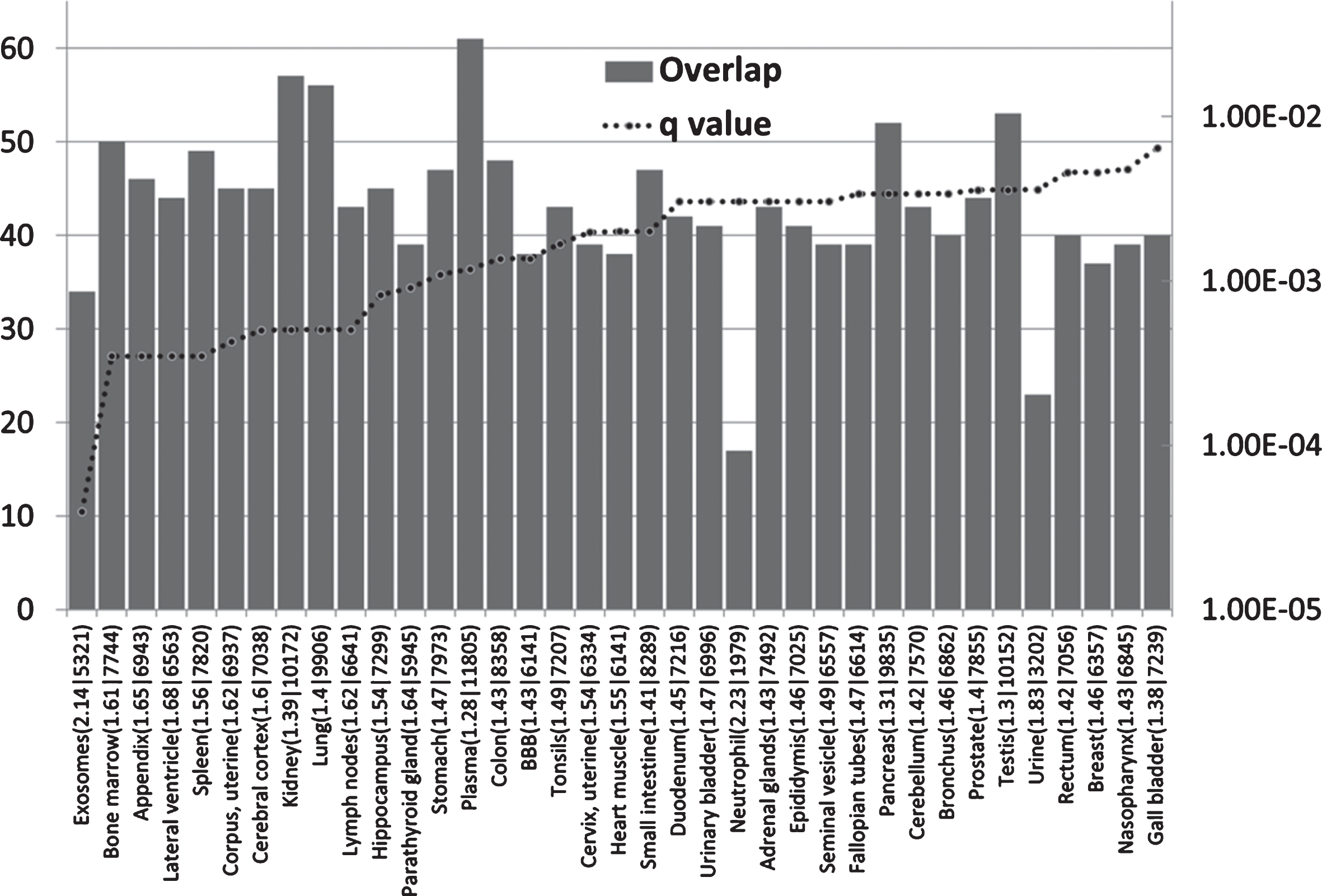
The distribution and enrichment of 78 AD genes in diverse proteomic and genomic datasets. The bars indicate the number of genes (from 78) in each tissue (Y1 axis) and the dotted line the FDR corrected p value (q value). The maximum on the Y2 axis is set to q = 0.05. Observed/expected values, followed by the total number of genes expressed per tissue, are appended after the identities of each sample on the X axis. BBB refers to a separate blood-brain barrier proteomics dataset.
AD genes are also enriched in the lateral ventricle, a site of the choroid plexus [119]. This provides CSF and is the location of the blood-CSF barrier, which is exploited by pathogens to gain access to the brain. The choroid plexus plays an important role in pathogen defense [120]. Postmortem gene expression studies of the choroid plexus epithelium in AD patients show changes indicative of increasedpermeability of the blood-CSF barrier and a reduction of macrophage recruitment [121], factors that would favor pathogen entry and reduce their phagocytosis by macrophages. The hippocampus bulges into the temporal horn of the lateral ventricle [122] and this area, a keystone of AD pathology, is thus in close proximity to a major site of cerebral pathogen entry. AD genes are also enriched in a separate BBB dataset from mouse cerebral arteries. This is discussed in greater detail below. Other barriers in intestinal and pulmonary tissues, also enriched in AD genes (Fig. 1), might also be considered as potential sites of pathogen entry. Immune systems play an important role at barrier interfaces [123].
Although AD genes are expressed in other sites, the main focus, in terms of enrichment, relates to immune and barrier systems.
Host/pathogen interactomes are enriched in AD genes (Fig. 2)
All host/pathogen interactomes, except those of the Borna virus, Ebola virus, and the HERV-W retrovirus were significantly enriched in AD genes (FDR p < 0.05). Pathogen burden (cytomegalovirus,HSV-1, B. burgdorferi, C. pneumoniae, and H. pylori) has been associated with AD [6] and the pooled interactomes of these five pathogens (3,922 host genes) were significantly enriched in AD genes (p = 7.3E-6).Given the variety of pathogens reported in AD brains, other cumulative effects might be expected for various pathogen permutations.
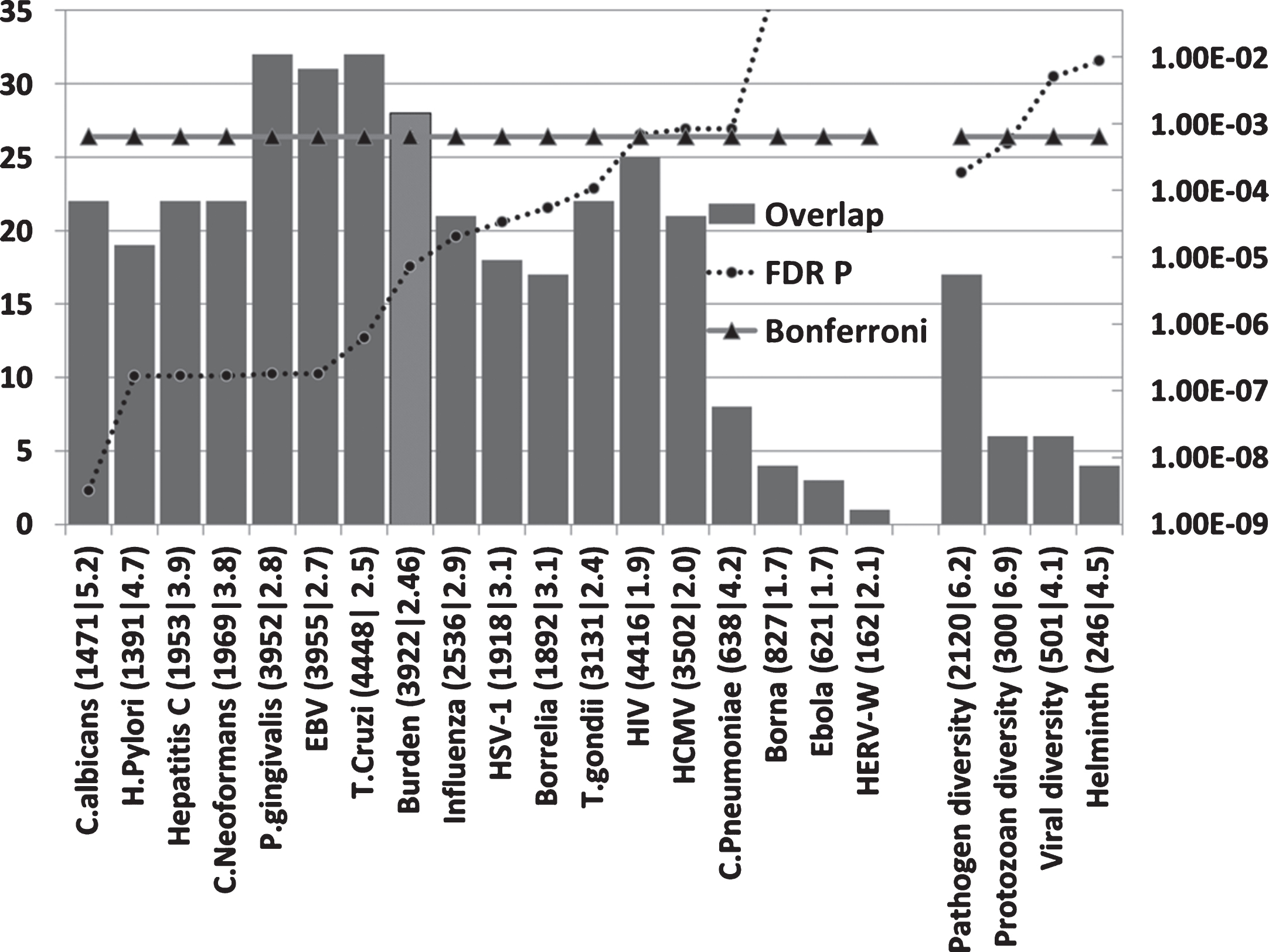
The number of AD genes (of 78) overlapping with diverse host/pathogen interactomes, or with those implicated in pathogen, protozoan, or viral diversity or with the immune response to parasitic worms (Helminth). The identities on the X-axis (e.g., C. albicans (1471|5.2) are appended with the total number of genes in each interactome (1471 in this case) or genetics dataset followed by the enrichment ratio (5.2 fold). The FDR-corrected p value for enrichment is shown on the right hand axis which is set to a maximum of 0.05. Invisible points are above this value. The Bonferroni cut-off level (p = 0.05/78) is also shown. The Burden data (lighter shaded bar) correspond to the combined interactomes of the human cytomegalovirus, HSV-1, B. burgdorferi, C. pneumoniae, and H. pylori.
The highest ranked pathogens in terms of the significance of the interactome/AD gene overlaps related to fungi (C. albicans and C. neoformans), the gum disease pathogen P. gingivalis and the EBV and hepatitis C viruses. Numerous fungal species, including C. albicans, have been detected in the AD brain, although C. neoformans was not one of the species studied. EBV has been associated with AD and hepatitis C associated with dementia (see Introduction). Several of these pathogens, including B. burgdorferi, P. gingivalis, H. pylori, influenza A virus, herpes virus, hepatitis C, cytomegalovirus, and HIV-1, or infectious burden (C. pneumoniae, H. pylori, cytomegalovirus, and HSV-1 and 2) have also been associated with atherosclerosis [124–126], an important endophenotype inAD [127].
Pathogen interactomes are enriched in the proteins found in AD amyloid plaques and neurofibrillary tangles (Fig. 3)
All pathogen interactomes were significantly enriched in proteins found in plaques and all except HERV-W and Burgdorferi interactomes significantly enriched in tangle proteins. The Borna virus and HIV-1 ranked highly in both cases. There is only one publication relating to Borna virus effects on Aβ and none could be found for tangles. The microglial activation produced by the virus reduced brain parenchymal, but increased cerebral vascular Aβ deposition, in AβPP transgenic mice [13]. The top agents relating to plaques were predominantly viral, while those relating to tangles were mostly viral, but included the parasites, T. cruzi and T. gondii.
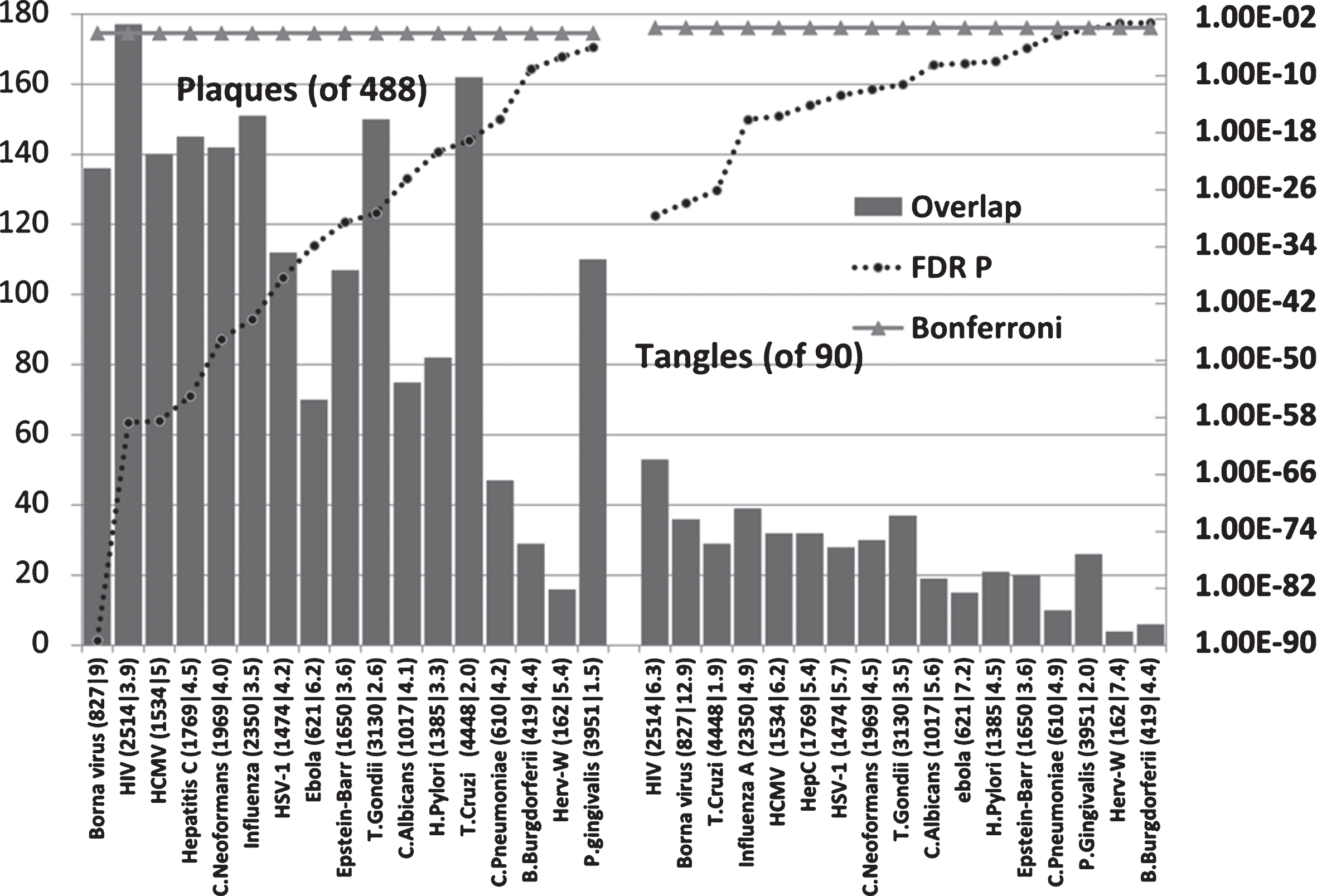
Host pathogen interactome enrichment in a set of 488 proteins isolated from amyloid plaques in the AD brain or from 90 proteins isolated from neurofibrillary tangles. Axes as for Fig. 2.
Aβ is an antibacterial, antifungal, and antiviral agent. It has been shown that it binds to C. albicans and S. typhimurium [47] and presumably to other microbes. Such microbes may well have sequestered host proteins specific to their particular life cycles during their passage to the cell, and this would partly explain the interactome enrichment. In addition to the plaque proteins relating to pathogen life cycles (for example receptor binding, endocytosis, and transport between intracellular compartments or nuclear entry and subsequent translation in the case of HSV-1), the proteins found in plaques and tangles contain many related to the immune system, inflammation and autophagy, all of which play a general role in pathogen defense [84–86] as does Aβ.
Viruses are transported via the microtubule network [128], which is also exploited by C. pneumoniae, T. cruzi, and T. gondii to reorganize cellular organelles to the pathogens’ advantage [129, 130]. Phosphorylated tau is a hallmark of neurofibrillary tangles and is induced by many pathogens. Tau inhibits the motility of the kinesin-1 motor protein that transports cellular organelles (or viruses) along the microtubules and tau phosphorylation counteracts this inhibition [131], but it is not clear whether this benefits or deleteriously affects the pathogens.
AD genes overlap with those implicated in pathogen, protozoan, or viral diversity or with the immune response to parasitic worms (Fig. 2)
The AD genes are enriched in a series of genome-wide and global-wide datasets related to general pathogen diversity, protozoan, or viral diversity (the number of different pathogens in a geographic region) or with the immune response to parasitic worms, most significantly so for general pathogen and protozoan diversity (FDR p < 0.05).
APOE4 is globally located in areas with a high parasitic burden, and is protective against virulent pathogens inducing malaria or hepatitis C (see Introduction) and these data show that several other AD genes relate to microbial diversity. In evolutionary terms, the maintenance of genes related to pathogen diversity in the population likely reflects a selective pressure for pathogen resistance, rather than susceptibility [92]. The AD genes common to these pathogen diversity datasets may thus be better equipped to deal with infections, although the mechanisms involved, including Aβ production and enhanced inflammatory responses may have deleterious cytotoxic effects if pathogens are able to infiltrate the brain. This is further discussed below.
Antimicrobial and immune/inflammatory gain of function in AD
The selection of AD genes for pathogen resistance implies that they must be better able to defend against pathogens. A high percentage of AD GWAS genes are involved in AβPP processing and the AD genetic variants favor an increase in Aβ production [78, 79]. Any genetic variant resulting in the increased production of this potent antimicrobial peptide could thus be considered as beneficial in evolutionary terms.
The APOE4 variant is also associated with enhanced immune/inflammatory responses. TLRactivation (TLR3, 4) in microglia induces cyclo-oxygenase-2 (PTGS2), microsomal prostaglandin E synthase (PTGES), and prostaglandin E2, an effect exaggerated in APOE4 (+/+) mice [132]. APOE4 is also associated with enhanced in vivo innate immune responses in human subjects. Whole blood from healthy APOE3/APOE4 volunteers induced higher cytokine levels on ex vivo stimulation with TLRs (TLR2, 4, or 5) ligands than blood from APOE3/APOE3 patients [133]. Gain of function also applies to AD variant forms of complement receptor CR1, which are better able to bind complement component C1q or C3B [134]. C1q and C3B are opsonins that interact with complement cell-surface receptors (C1qRp, CR1, CR3, and CR4) to promote phagocytosis (including that of infectious agents) and a local pro-inflammatory response [135]. TREM2 variants in AD are also associated with enhanced inflammatory responses (upregulation of proinflammatory cytokines) [136]. In presenilin (PSEN1) mutant knockin mice, microglial challenge with bacterial LPS results in enhanced nitric oxide and inflammatory cytokine responses, relative to normal mice [137]. For these genes at least, this gain of immune/inflammatory function concords with selection for pathogenresistance.
It has also been noted that unaffected offspring with a parental history of AD have an enhanced inflammatory response in LPS-stimulated whole blood samples, producing higher levels of interleukin 1 beta, tumor necrosis factor alpha, and interferon gamma in response to LPS. This effect was independent of the APOE4 variant [138] suggesting that other AD genes are also endowed with gain of function in relation to the immune/inflammatory system. Monocyte-derived dendritic cells from AD patients also produce more interleukin-6 than those from healthy controls. AD monocytes stimulated with LPS also show a higher induced expression of the pro-inflammatory ICAM1 adhesion molecule than controls [139]. Aβ also stimulates cytokine production in peripheral blood mononuclear cells (PBMC) and the production of the chemokines, RANTES, MIP-1beta, and eotaxin as well as that of CSF2 (colony stimulating factor 2 (granulocyte-macrophage)) and CSF3 (colony stimulating factor 3) is greater than controls in AD-derived PBMC stimulated withAβ [140].
Inflammation, the inflammasome, and other pathogen-defense mechanisms, for example free radical generation, are themselves neurotoxic and their contribution to neuronal cell death in AD isincreasingly recognized [141–146].
In summary, AD susceptibility genes appear to have been subject to positive selective pressure. This may relate to their ability to convey general resistance to dangerous pathogens over evolutionary time, and this may in turn relate to enhanced immune/inflammatory responses to pathogens. Many AD gene variants converge towards an enhanced production of Aβ, and as this peptide has potent broad-spectrum antimicrobial effects, this may also confer an evolutionary advantage.
Host/pathogen interactome enrichment in misregulated genes of the AD hippocampal transcriptome (Fig. 4)
All pathogen interactomes, most notably relating to influenza, C. neoformans, and Hepatitis C were highly enriched in genes relating to this microarray dataset (combined up and downregulated hippocampal genes in AD). The significance level of the interactome enrichment for most pathogens was several orders of magnitude below the Bonferroni cut off (p = 1.74E-05) (Fig. 4). Regarding the influenza data, bronchopneumonia, often caused by influenza, is a common final cause of death in dementia patients [147] and such recent infections close to death may well influence the data.
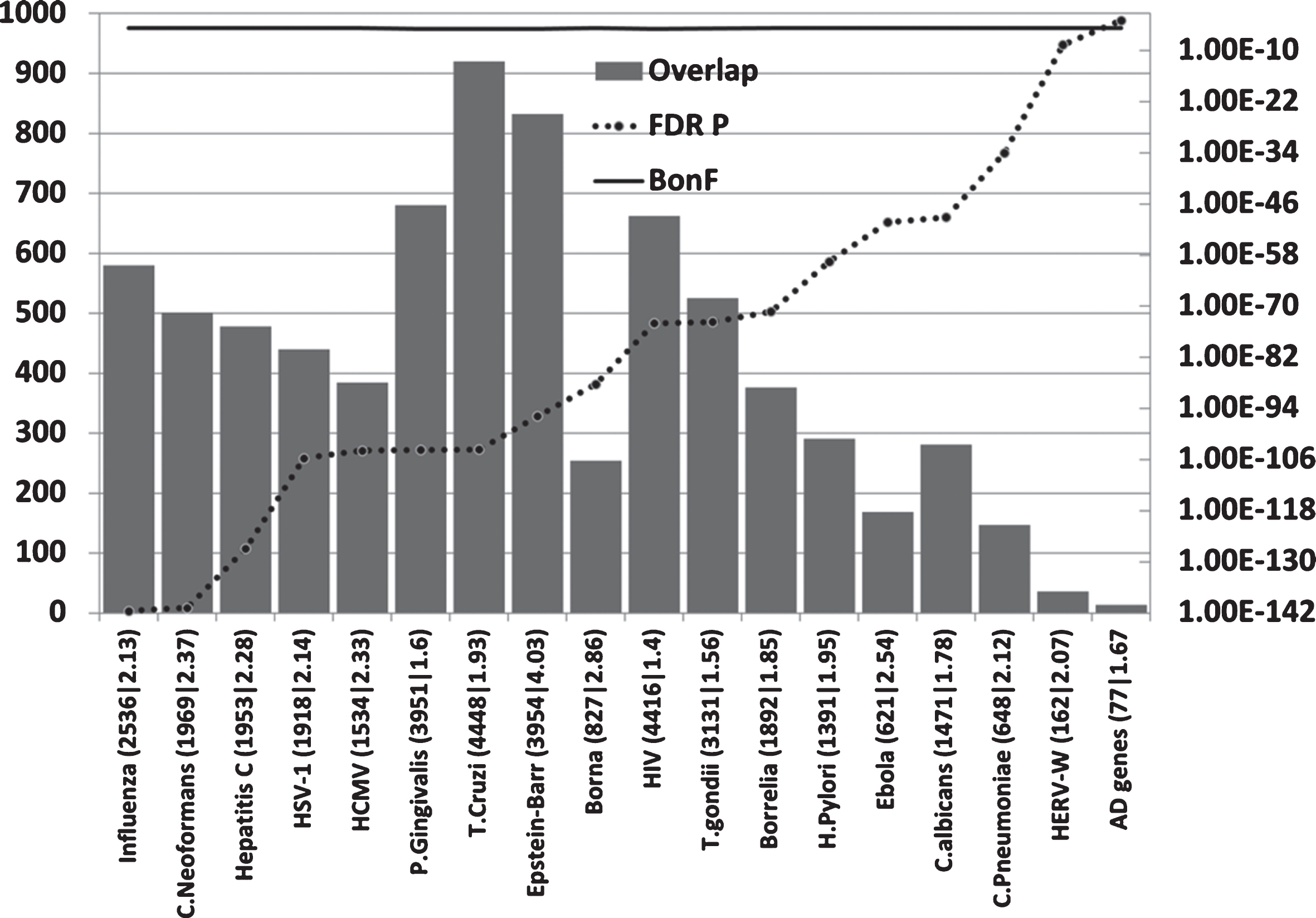
Overlaps between misregulated AD hippocampal genes (combined up and down) and host/pathogen interactomes. Axes as forFig. 2.
Regardless of the rank order, it is clear that many diverse pathogen interactomes affect several hundred genes of the 2,879 misregulated in the AD hippocampus and/or that these misregulated AD genes represent a substantial percentage of the individual pathogens’ interactomes (Fig. 4).
Kegg pathway analysis of these misregulated genes using the Consensus Path Database [148] showed that many infection-related pathways were also significantly enriched in the hippocampal dataset (FDR p < 0.05). These included (pathogen with N genes common to the pathogen pathway and the hippocampal AD microarray; followed by the FDR corrected p value): EBV infection (74,5.5E-7); Salmonella infection (36,0.0001); Tuberculosis (57,0.0009); Epithelial cell signaling in H. pylori infection (28,0.00097); Shigellosis (27,0.001); Influenza A (54,0.003); Herpes simplex infection (56,0.0036); Vibrio cholerae infection (21,0.0089); HTLV-I infection (71,0.0096); Toxoplasmosis (37,0.013); Hepatitis B (43,0.018); Pathogenic Escherichia coli infection (20,0.02); Bacterial invasion of epithelial cells (26,0.02); Measles (38,0.04).
Upregulated genes in the AD hippocampus are enriched in genes upregulated by viral, bacterial and fungal pathogens or TLR ligands
Most MSigDB or GEO microarray experiments related to immunocompetent blood cells (B cells,T cells, dendritic cells, monocytes, and macrophages) or from cultured cell lines. Upregulated genes from these datasets were compared with the upregulated genes (n = 1,690) from the AD hippocampal study, which contain the pathways relevant to pathogens and immune activation (inflammation, complement activation, and the defense response) [83].
The hippocampal genes upregulated in AD were significantly enriched in upregulated genes in datasets from multiple viral species and responses to double stranded RNA or to the double-stranded RNA viral mimic/TLR3 agonist, Polyinosinic:polycytidylic acid (poly I:C) (Fig. 5). The viruses ranged from the benign (e.g., the rhinovirus that causes the common cold) to the highly malignant (e.g., the ebolavirus, rabies virus or HIV-1). They include common human infectious agents (e.g., adenovirus 5, influenza, EBV, HSV-1, measles or the Norwalk virus). Apart from HSV-1, the human cytomegalovirus, HIV-1, or hepatitis C, none of these have been implicated in AD or dementia. No infection-related datasets were found for microglia, the brain resident immunocompetent cells, but significant enrichment of the AD upregulated genes was observed for genes upregulated by interferon gamma in microglial cells (Fig. 5). Interferon gamma plays an important role in the response to viral, bacterial, and parasitic infections [149].
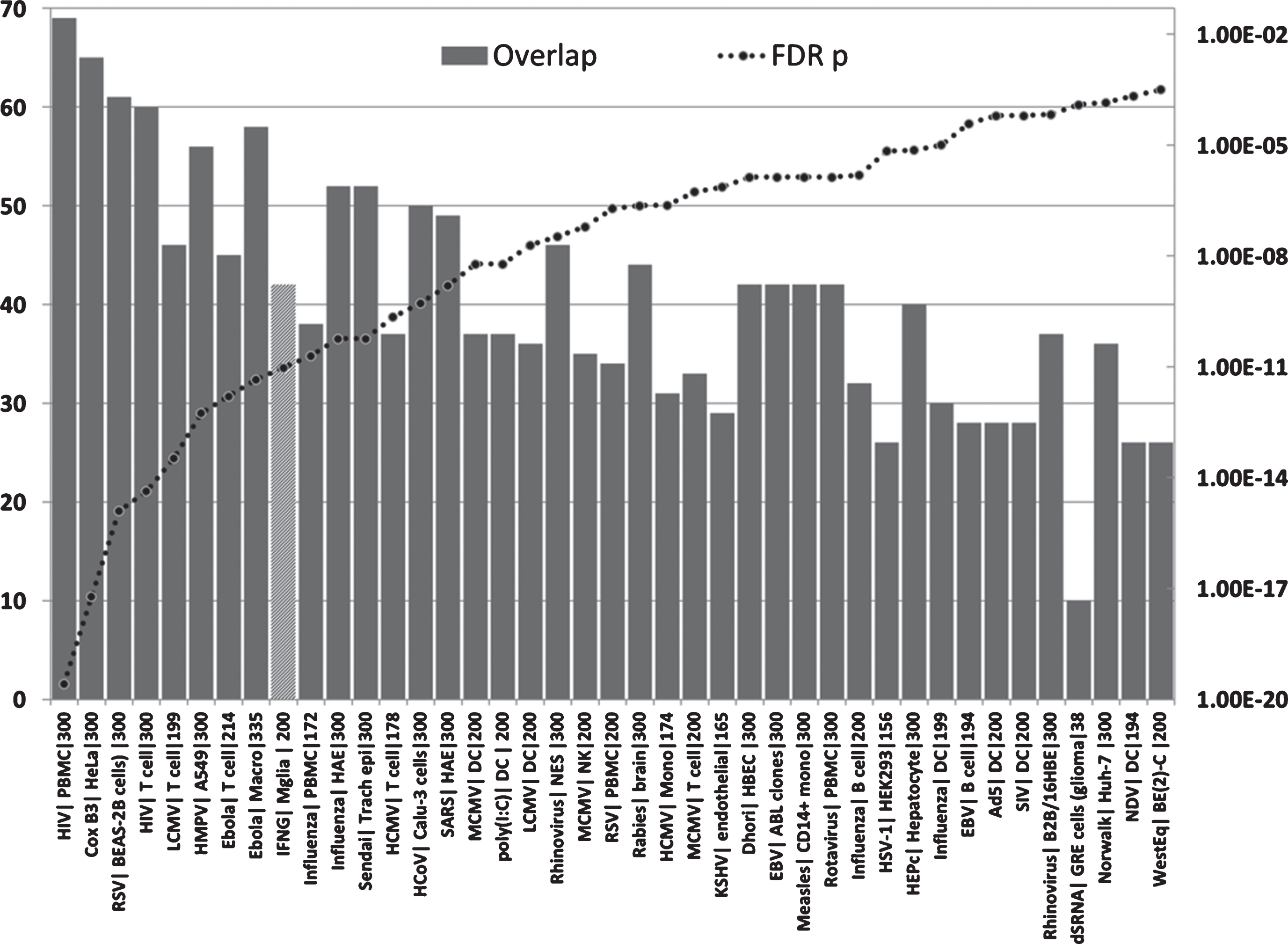
Overlaps between genes upregulated in the AD hippocampal transcriptome and those upregulated by viruses, interferon gamma or the viral mimic poly(I:C) in immunocompetent cells (MSigDB or GEO data). (X axis = virus| celltype|N upregulated genes from MSigDb or Geo): Y1 = N overlap; Y2 = p value. Tissue/cell abbreviations: A549, adenocarcinomic human alveolar basal epithelial cells; ABL, Akata Burkitt’s lymphoma cells; B2B/16HBE, BE(2)C or BEAS-2B, human bronchial epithelial cells; BroLav, human bronchial lavage; Calu-3, cultured human airway epithelial cells; DC, dendritic cells; GRE, glioma cell line; HAE, human airway epithelial cells; HBEC, human bronchial epithelial cells; HEK, human embryonic kidney cells; HeLa, cervical cancer cell line; HuH-7, hepatocarcinoma cell line; Macro, macrophage; Mgli, microglia; Mono, monocytes; NES, human nasal epithelial scrapings; NK, natural killer cells; PBMC, peripheral blood mononuclear cells; PLC/PRF/5 cells, human liver hepatoma cells; Trach epi, Tracheal epithelial cells. Viral abbreviations: HIV, human immunodeficiency virus; Cox B3, Coxsackie B3 virus; RSV, respiratory syncytial virus; LCMV, lymphocytic choriomeningitis virus; HMPV, human metapneumovirus; Ebola, Ebola virus; Influenza, Influenza A virus; Sendai, Sendai virus; HCoV, human coronavirus; IFNG, interferon gamma; SARS, severe acute respiratory syndrome coronavirus; HCMV, human cytomegalovirus; MCMV, mouse cytomegalovirus; Dhori, Dhori virus; EBV, Epstein-Barr virus; HepC, hepatitis C virus; KSHV, Kaposi’s sarcoma-associated herpesvirus; HSV-1, herpes simplex; Norwalk, Norwalk virus (Norovirus); Ad5, adenovirus 5; SIV, Simian immunodeficiency virus; poly(I:C), Polyinosinic:polycytidylic acid (a viral mimic stimulating TLR3 receptors); NDV, Newcastle disease virus; WestEq, Western equine encephalomyelitis virus; LASV, Lassa virus; dsRNA, double stranded RNA; HEV, hepatitis E virus.
The upregulated hippocampal genes in AD were also enriched in infection datasets for numerous bacteria as well as to fungi (C. albicans and C. neoformans) and in those related to bacterial endotoxin or sepsis and to nematode/trematode or protozoan infection datasets (FDR p < 0.05) (Fig. 6). This also applied to diverse LPS datasets and responses to TLR ligands, CpG oligonucleotide (a ligand for TLR9, which mediates cellular response to unmethylated CpG dinucleotides in bacterial DNA or R848 (a ligand for TLR7/TLR8 both of which recognize RNA released from pathogens that enter the cell by endocytosis [150]) (Fig. 6). With the exception of H. pylori, P. gingivalis and B. burgdorferi, and C. albicans or C. Neoformans, none have been implicatedin AD.
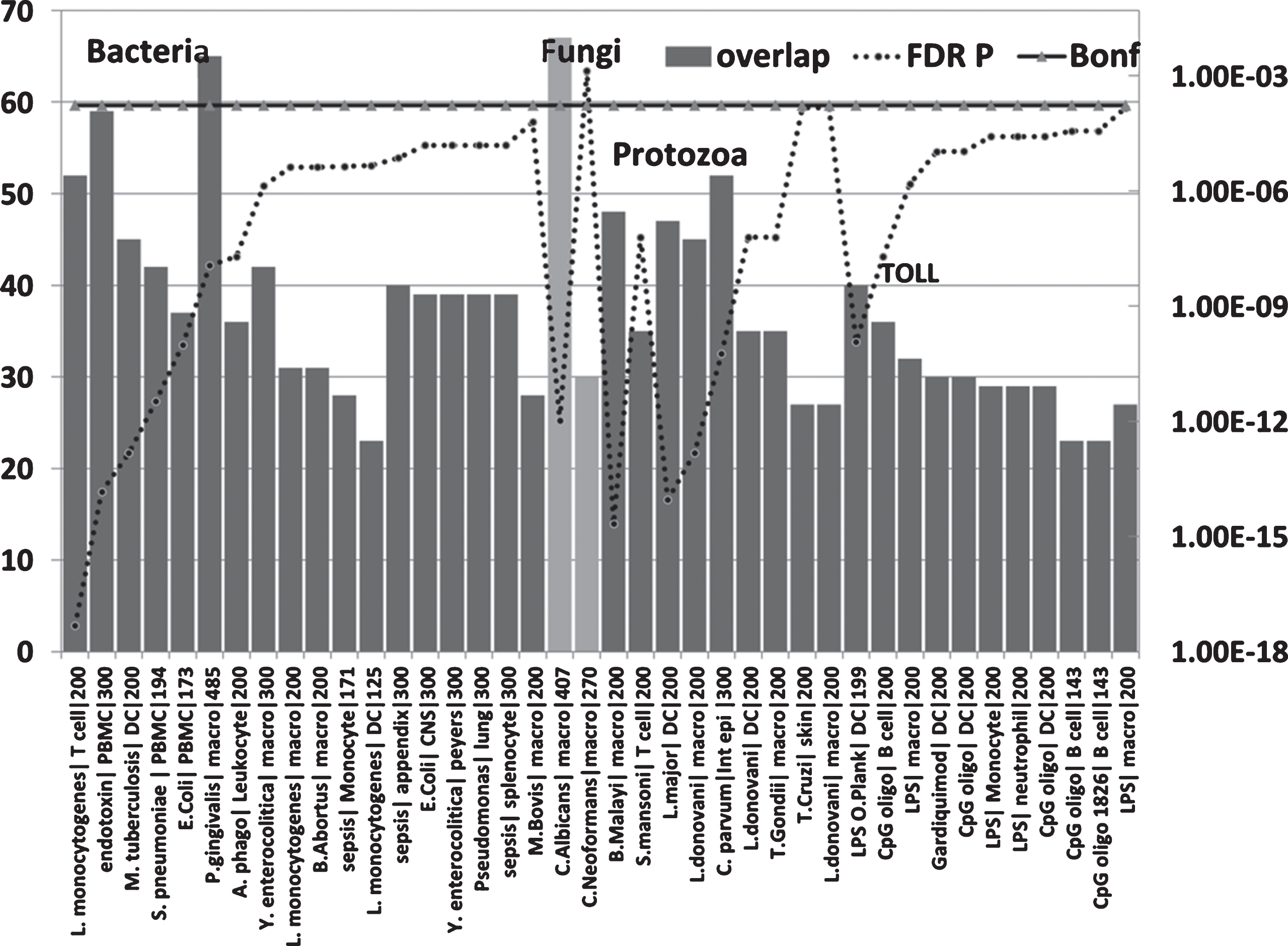
Overlaps between genes upregulated in the AD hippocampal transcriptome and those upregulated by bacteria, fungi, nematode (B. malayi)/trematode (S. mansoni), protozoan, LPS and TLR ligands in immunocompetent cells (MSigDB or GEO data). (X axis = pathogen/ligand| celltype|N upregulated genes from MSigDb or Geo): Y1 = N overlap; Y2 = p value). Pathogen or ligand abbreviations (from left): L. monocytogenes, Listeria monocytogenes; endotoxin, gram-negative bacterial wall component; S. pneumoniae, Streptococcus pneumoniae; E. coli, Escherichia coli; P. gingivalis, Porphyromonas gingivalis; A. phago, Anaplasma phagocytophilum; Y. enterocolitica, Yersinia enterocolitica; M. bovis, Mycobacterium bovis; C. albicans, Candida albicans; C. neoformans, Cryptococcus neoformans; B. malayi, Brugia malayi (filarial parasite causing elephantiasis); S. mansoni, Schistosoma mansoni; L. donovani, Leishmania donovani; C. parvum, Cryptosporidium parvum; L. major, Leishmania major; T. gondii, Toxoplasma gondii; T. cruzi, Trypanosoma cruzi; LPS, lipopolysaccharide; LPS O.Plank, Oscillatoria Planktothrix (cyanobacterial lipopolysaccharide); CpG oligo, CpG Oligodeoxynucleotide (TLR9 ligand); Gardiquimod, TLR7 ligand. Cell type abbreviations as for Fig. 5: + CNS, central nervous system; peyers, peyers patch; Int epi, intestinal epithelial cells.
Diverse pathogen sensors and defenders relating to bacteria, viruses, parasites, or fungi are upregulated in the AD brain, blood, or CSF
The data presented above shows that the hippocampal transcriptome in AD matches that induced by numerous pathogens. The misregulated genes include many related to inflammation, complement activation and the immune system that could relate to many pathogens but also to other neurotoxic promoters. Within the immune system there is a specific branch that is more specifically dedicated to pathogens. This involves pattern recognitionreceptors, sensors that detect viral DNA or RNA, and antimicrobial defensins, inter alia. These sense cell wall components of bacteria, fungi, or protozoa (TLRs, C-type lectin receptors, and NOD-like) [151, 152]. Others are designed to sense viral DNA or RNA. These include intracellular TLRs (TLR3,8,9), EIF2AK2 (eukaryotic translation initiation factor 2 alpha kinase 2), often known as pkr, RIG-I (coded by retinoic acid-inducible gene 1 = DDX58), MDA5 (Melanoma Differentiation-Associated protein; coded by IFIH1) and LGP2 (coded by DEXH-box helicase 58 = DHX58) [150, 154].
Indoleamine 2,3-dioxygenase 1 (IDO1) diverts tryptophan metabolism to N-formyl-kynurenine; (away from serotonin production). IDO1 upregulation is an important defense mechanism against pathogenic bacteria, many of which rely on host tryptophan. This IDO1 response is also deleterious to other pathogens and parasites, including T. gondii, and to a number of viruses, including HSV-1 and other herpes viruses. Quinolinic acid produced by IDO1 activation is neurotoxin acting via N-methyl-D-aspartate receptors [155]. Kynurenine and kynurenic acid, also produced by IDO1 activation, are ligands for the aryl hydrocarbon receptor, which plays an important role in antimicrobial defense and immune regulation [156]. In relation to viruses, the oxysterols 25-hydroxycholesterol and 27-hydroxycholesterol have also recently emerged as broad-spectrum antiviral agents inhibiting the replication of enveloped and non-enveloped viruses [157]. Sirtuins (SIRT1-7) also have broad spectrum antiviral activity against DNA and RNAviruses [158].
To a certain extent, each of these sensors and defenders show a degree of selectivity for viral, bacterial, fungal or protozoan infections, although with some overlap. A focus on this particular pathogen-specific area in relation to AD should thus be more indicative of the type of infection involved.
To this end, the pathogen specificity of these sensors and defenders was reviewed, generally and in relation to the pathogens in this study. These properties were then matched to reported expression changes in the AD brain, blood, or CSF in relation to the pathogens in this study and to others. This analysis is reported in Supplementary Table 2, which shows that the expression changes seen in AD relate tosensors and defenders for the diverse pathogens, and to many other viruses, bacteria, or fungi. They are not restricted to viral, fungal, or bacterial defense, supporting a polymicrobial involvement.
99 antimicrobial/pattern recognition, antiviral and related defensive genes were identified that were upregulated in the AD brain, blood, or CSF. No single pathogen or group of pathogens (bacteria, viruses, or fungi) accounted for all of these changes. In terms of the pathogens related to this study, influenza,C. albicans, and HSV-1 matched the highest number of AD-affected genes, but high scores were also obtained for other bacteria (including Enterococci, E. coli, streptococci, staphylococci, pseudomonas, listeria), fungi (including Aspergillus and Saccharomyces species), or viruses (e.g., Sendai virus, coxsackie B virus, coronaviruses, and flaviviruses (dengue virus and West Nile virus) (Supplementary Table 2: Fig. 7A).
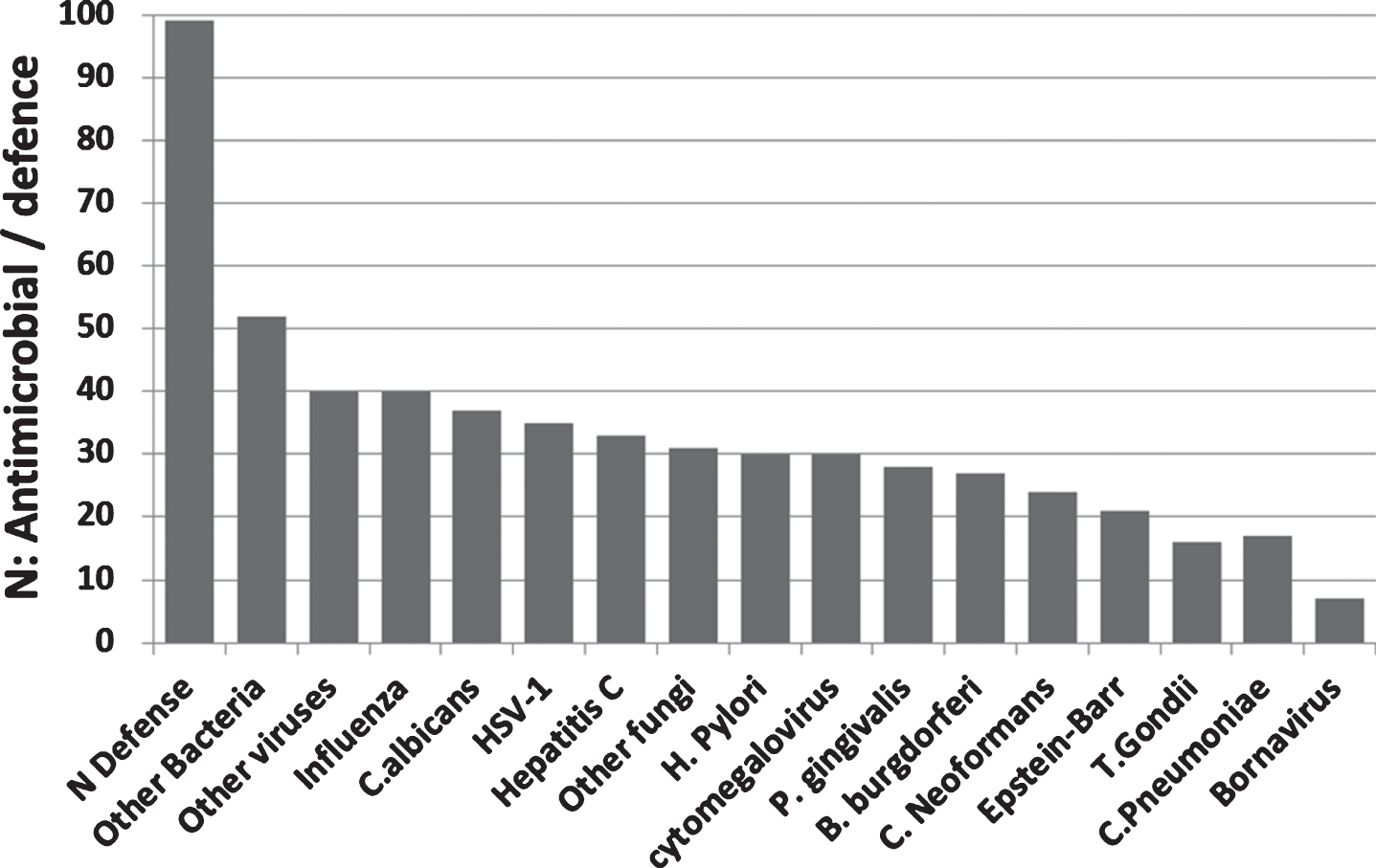
(A) The number of pathogen defense related genes or proteins (of 99 mostly upregulated = N defense) in the AD brain, blood, or CSF, plotted in relation to the known matched effects of the pathogens studied and to general bacterial, viral or fungal defense (Other). These are referenced in Supplementary Table 2 and include antimicrobial peptides, pattern recognition receptors, interferons, antiviral effectors, complement related, Fc gamma receptors, and other dedicated pathogen detection/defenseprocesses.
The relative selectivity relating to three pathogens that have been detected in the AD brain (the HSV-1virus, fungal C. albicans, and the spirochete B. burgdorferi) is shown in the Venn diagram (Fig. 7B). From this analysis, it appears that there are a number of misregulated sensors and defenders in AD that are both promiscuous (reacting to many species) and specific, showing a relative preference for these viral, fungal or bacterial pathogens.
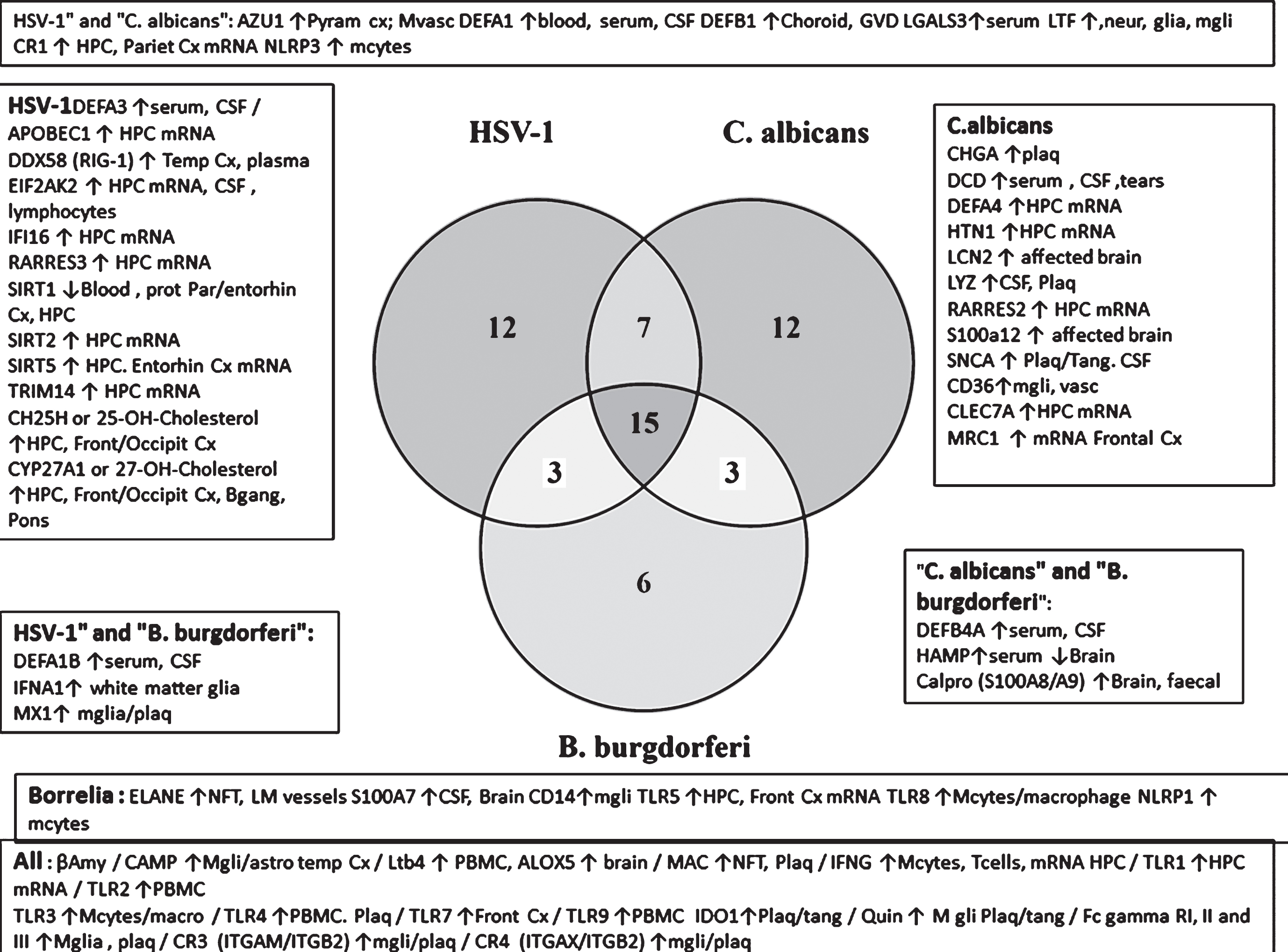
(B) Defenders and sensors and their expression (↑= upregulated; ↓= downregulated) in the brain, blood, or CSF of Alzheimer’s disease patients. These effects are matched to the effects of HSV-1, C. albicans, or B. burgdorferi as viral, bacterial and fungal representatives. These effects and those related to other pathogens are referenced in Supplementary Table 2. Regions: CP, choroid plexus; Cortical areas (Cx, cortex; Entorhin, entorhinal; Fr, frontal; par, parietal; Temp, temporal; occip, occipital); CSF, cerebrospinal fluid; GVS, granulovacuolar degeneration; HPC, hippocampus; lympho, lymphocytes; macro, macrophages; mcyt, monocytes; mgli, microglia; Mvasc, microvasculature (brain); Neur, neurons; NFT, neurofibrillary tangles; PBMC, peripheral blood mononuclear cells; Plaq, amyloid plaques; Ser, serum; tang, tangles. Antimicrobial peptides and related: AZU1, azurocidin; βamy, amyloid-β; APCS, amyloid P component, serum; Calpro, calprotectin (= S100A8+S100A9); CHGA, chromogranin A; DCD, dermcidin; αdefs or βdefs, unspecified alpha or beta defensins: CAMP, cathelicidin antimicrobial peptide (LL-37); CHI3L1, chitinase 3 like 1 (aka YKL-40); Defensins, DEFA’s, DEFB’s: ELANE, elastase; HAMP, hepcidin; HTN1, histatin 1; IAPP, islet amyloid polypeptide (Amylin); LCN2, lipocalin 2; LTF, lactotransferrin; S100’s, S100 calcium binding protein; SNCA, alpha synuclein. Pattern recognition receptors: CD14; CD36; C-type lectin, CLECs; MRC1, mannose receptor C type 1; Toll-like receptors, TLR1 to 10. Viral detectors and antiviral: DEAD box proteins, DDXs; EIF2AK2, eukaryotic translation initiation factor 2 alpha kinase 2 (pkr); SIRTs, sirtuins; 25- and 27-OH-cholesterol, 25- or 27-hydroxycholesterol synthesized by CH25H (cholesterol25- hydroxylase) and a cytochrome p450 enzyme (CYP27A1); IFI16, interferon gamma inducible protein 16; MX1, MX dynamin like GTPase 1; TRIM, tripartite motif protein; Complement related: CR1, complement receptor 1; CR2, complement receptor 2 (integrins: ITGAM/ITGB2); CR4, complement receptor 4 (Integrins: ITGAX/ITGB2); MAC, membrane attack complex (complement components C5b-C9). General: AGER, advanced glycosylation end product-specific receptor (also known as RAGE); CRP, C-reactive protein; Fc Gamma R, Fc gamma receptors; IDO1, indoleamine 2,3-dioxygenase 1; Interferons, IFNA1, IFNA5, IFNB1, IFNG; Integrins, ITGAM/ITGAX/ITGB1; LGALS3, lectin, galactoside binding soluble 3; LTB4, leukotriene B4, synthesized by ALOX5); NLRP1, NLR family pyrin domain containing 1; Quin, quinolinic acid; RARRES2 and 3, retinoic acid receptor responder (2 and 3).
The promiscuous defensive agents include antimicrobial agents (Aβ and cathelicidin/LL-37), leukotriene B4 and its synthesizing enzyme 5-lipoxygenase (ALOX5), complement receptors (CR3, CR4), and the membrane attack complex, Fc gamma receptors, interferon-gamma and TLRs (TLR1, 2, 3, 4, 7, and 9). Indoleamine dioxygenase (IDO1) and its derivative, the neurotoxic quinolinic acid also belong to this group. More selective antiviral agents (HSV-1) include the oxysterols, 25- and 27-hydroxycholesterol and their synthesizing enzymes (CH25H and CYP17A1), DDX58/RIG-1, EIF2AK2, and sirtuins. Those related to C. albicans include the C-type lectin CLEC7A, the mannose receptor MRC1, CD36, and the antimicrobial peptides, chromogranin A, dermcidin, histatin, and synuclein. Relatively selective spirochete defense is exemplified by neutrophil elastase, CD14, and TLRs (TLR5, TLR8). This analysis suggests that the antimicrobial alerts in AD reflect a polymicrobial origin. It should, however, be recognized that such specificity is not absolute for other viral, fungal, or bacterial species. For example, in relation to B. burgdorferi selective agents, TLR5 also recognizes bacterial flagellin (Refseq) and TLR8 recognizes single stranded viral RNA [159]. Alpha-synuclein(C. albicans-selective) has antifungal effects, and is also antibacterial (Escherichia coli and Staphylococcus aureus) [160].
These data (Supplementary Table 2, Figs. 5–7) show that upregulated AD hippocampal genes match those upregulated by multiple pathogens in immunocompetent cells and that sensors and defenders relating to multiple pathogens are upregulated in the AD brain, blood, or CSF. These involve reactions to many of the pathogens of this study, representing many different classes (bacteria, viruses, fungi, and parasites) and there appears to be no discrimination, or focus on any particular type. This would concord with the multiple and diverse pathogen species that have been detected in the AD brain and with the relationship between the AD genes or the hippocampal transcriptome with multiple pathogen species. In this analysis, a variety of pathogens were used based mostly on their relationship with AD epidemiology, but also to include diverse families (viruses, bacteria, fungi, and protozoa). Those relevant to most AD cases will presumably relate to the most common infections in the elderly. In the US, these include influenza, bacterial pneumonia, urinary tract infection, intra-abdominal infections, gram-negative bacteremia, and infection of pressure ulcers[161, 162]. These are caused by more common bacteria, fungi, and viruses, rather than by more exotic species such as the Bornavirus or T. cruzi. Despite the potential relationship of so many pathogens to AD, their cerebral entry will still also depend upon on the multiple and manageable environmental factors that disrupt or prevent BBB malfunction. Until more extensive studies of the blood and brain microbiomes in AD, the relative importance of individual or multiple species can only be speculative. A recent investigation of the gut bacterial microbiome in elderly cognitive impairment, associated with brain amyloidosis, reported an increase in the abundance of pro-inflammatory Escherichia/Shigella taxa and a reduced abundance of an anti-inflammatory taxon; E. rectale. These changes were associated with a pro-inflammatory blood cytokine profile in subjects with brain amyloidosis [163], but to date there have been few brain microbiome studies in AD, or studies of the peripheral AD virome orfungome.
Together these data suggest a significant parallel between the upregulated genes in the AD hippocampus and the responses to multiple and diverse infectious agents with little overall discrimination between viral, bacterial, fungal, or protozoan types of infection. This diversity is supported by the pattern recognition and antimicrobial survey. Multiple pathogens have been detected in the AD brain and the diversity of these infection related overlaps with the AD hippocampal transcriptome suggests that many other pathogens could induce similar pathological transcriptome changes. Microbiome studies in the AD brain and periphery will help to elucidate the role of multiple pathogens.
AD genes are localized in the BBB
30/78 AD genes are expressed in the BBB proteome dataset of mouse cerebral arteries [101] (Fig. 1). The list below indicates the 30 BBB genes, annotated with the number of pathogen interactomes with which they overlap. Most BBB expressed genes interact with none or few pathogens (5 or less of the 17 studied), suggesting a subdivision of mainly BBB and mainly pathogen related. This might be confounded by missing data, as several of these genes are poorly characterized in terms of function. These 30 genes (N interactomes in brackets) are: PCNX (0), ABCA7 (1), ADAMTS20 (1), ATXN7L1 (1), TREML2 (1), AP2A2 (2), BCAM (2), CNTNAP2 (2), ECHDC3 (2), FRMD4A (2), GRIN3B (2), PAX2 (2), PICALM (2), DISC1 (3), LUZP2 (3), RELN (3), TTLL7 (3), FERMT2 (4), HMHA1 (4), MSRA (4), PPP1R3B (4), SASH1 (4), BIN1 (5), SORL1 (5), PVRL2 (7), MMP12 (8), CLU (9), PTK2B (10), BCL3 (13), SQSTM1 (13). CD2AP (7 interactomes), not present in the mouse cerebral arteries dataset, has also been shown to be involved in the maintenance of BBB integrity [164].
The BBB location of a high proportion of AD genes indicates an important function in relation to AD.
Routes of pathogen entry to the brain: olfactory routes and control of the BBB
The pathogen data presented above highlight multiple relationships between AD and diverse pathogens. Aβ production can also now be viewed as an innate immune response to pathogen attack. These data show that many pathogens cause Aβ deposition and that they play a major role in AD pathogenesis.
Nevertheless, pathogens have to enter the brain, via the BBB or other routes, to evoke such changes and the old age of AD patients suggests that age must be a major determinant of such effects. The effects of aging in relation to the BBB and the immune system and to routes of pathogen entry are discussed below, together with other environmental risk factors that affect BBB integrity.
BBB breakdown in aging and its control by the basal forebrain and locus coeruleus
Aging itself results in a loss of BBB integrity [165] and in immune function (immunosenecence) [31] and these two factors would be expected to favor blood-borne pathogen survival and cerebral pathogen entry. In normal human aging, loss of barrier integrity appears to begin in the hippocampus, a crucial area in AD and in learning and memory [166]. The BBB consists of a series of capillaries, capillary arterioles, and capillary venules serving neurons throughout the brain. This network of fine blood vessels, which shows early pathological changes in AD, is in turn controlled by a perivascular neuronal plexus originating mainly from the locus coeruleus, the nucleus basalis of Meynert, and the basal forebrain area, each of which suffer early extensive damage in AD [167]. A recent MRI study suggests that degenerative changes in the basal forebrain including the nucleus basalis of Meynert are early events in AD that precede cortical degeneration [168]. Degeneration of the cortical cholinergic innervation originating from the basal forebrain was one of the first neurochemically characterized lesions in AD [169, 170]. The basal forebrain cholinergic system receives many olfactory inputs from the olfactory bulb, anterior olfactory nucleus, and the pyriform cortex [171]. Damage to the olfactory bulb and the olfactory tracts and system are also early characteristic features of AD[172, 173]. Anosmia/olfactory impairment are early and predictive signs of mild cognitive impairment and progression to AD [174] and Aβ deposition and tau pathology can be observed in the nasal epithelium in the majority of AD cases [175].
Use of the olfactory route to the basal forebrain and locus coeruleus by herpes simplex and other pathogens
Many viruses including Herpes viruses (HSV-1 and HHV6), the Borna disease virus, rabies virus, and the influenza A virus are known to use the olfactory route to invade the brain [14]. It has also been shown in mice that injection of HSV-1 into the olfactory bulbs leads to viral migration into the amygdala and hippocampus via the olfactory nerve and the locus coeruleus [176]. Nasal inoculation with C. pneumoniae in mice also results in cerebral Aβ deposition suggesting use of the olfactory route [118]. The nasal epithelial mucosa forms an important physical and immune barrier to invading pathogens [177, 178]. While immunosenescence occurs in many mucosal sites during aging, nasopharyngeal tissue function is more resistant and remains intact during aging with notable signs of immunosenescence seen only in the very elderly [179]. The nasal route of pathogen entry would thus also be restricted to the aged population.
Damage to the basal forebrain including the nucleus basalis of Meynert is also a feature of Herpes simplex encephalitis in Man [180] and HSV-1 (or vaccinia) infection in mice results in increased vascular permeability in the olfactory bulb, hippocampal regions and in areas surrounding the cerebral ventricles [26]. Both the noradrenergic locus coeruleus [181, 182] and the cholinergic connections from the basal forebrain [183–186] control the cerebral vasculature, cerebral blood flow and vascular permeability. It is thus conceivable that pathogen related damage to the basal forebrain/locus coeruleus contributes to subsequent cerebrovascular and BBB defects and to the sequential pathological anatomy in AD. Aβ itself can produce BBB damage [187]. This could create a feed-forward effect whereby pathogen-induced Aβ production produces further BBB damage, spreading from the pathogen transit zones.
Some pathogens can also circumvent the BBB by direct attack. For example, certain strains of neuroinvasive B. burgdorferi are known to be able to translocate across the BBB initially via anchoring to host proteins on brain microvascular endothelial cells and subsequently via activation of metalloproteases that degrade extracellular matrix and intercellular junctions [28]. C. pneumoniae infects blood vessels and monocytes in the AD brain, and also promotes the invasion of the infected monocytes through human brain endothelial cells indicating BBB breakdown [29]. The fungal pathogen C. neoformans uses a Trojan horse strategy where infected phagocytes cross brain microendothelial barriers [30]. This pathogen may also actively disrupt BBB function be secreting fungal proteases [188, 189]. P. gingivalis also disrupts epithelial barrier function via secreted proteases (gingipains) that degrade cadherins (CDH1, CDH2,CDH5) as well as occludin, beta- and gamma– catenins, paxillin, and integrins (ITGA3, ITGB1, ITGB4) [190–192]. Gingipains have been detected in the hippocampal microvasculature of P. gingivalis-infected APOE (– /– ) mice and likely contribute to impaired BBB integrity and to the cerebral invasion of P. gingivalis in this model [27]. The prevalence of periodontitis increases with age [193]; this is again a factor that would limit effects on BBB function to the aged. Any pathogen that so disrupts the BBB might be expected to influence invasion by others.
In addition the BBB is disrupted by the systemic administration of bacterial LPS [24] or by lipoteichoic acid, a cell wall component of gram-positive bacteria [25] suggesting that repeated peripheral bacterial infections (a feature of the elderly population) could also weaken the BBB.
Immunosenescence in aging
Aging-related immunosenescence can increase susceptibility to pathogens due to immunodeficiency, but it is also accompanied by an increase in the pro-inflammatory activity of monocytes and macrophages which can lead to chronic low grade inflammation, termed “inflamm-aging” [31, 32]. This increased inflammatory function also applies to microglia [194]. Certain AD gene variants are associated with enhanced pro-inflammatory responses (see above) and cerebral pathogen entry would thus be met with a doubly vigorous inflammatory response related to both immunosenescence and genetic variation. Persistently activated monocyte/macrophages have been observed in the blood of patients with early AD [195] and increased activation of microglia/macrophages, colocalized with the area of heavy Aβ concentration, is also observed in the brains of AD patients [196].
Abrogation of the peripheral antimicrobial effects of Aβ by autoantibodies?
The antimicrobial effects of Aβ are blocked by an antibody to this peptide [48]. A number of studies have reported the presence of autoantibodies to Aβ in the serum of the elderly [197] and in AD [198, 199].They are also observed in mild cognitive impairment patients with higher plasma levels in those who progressed to AD [200]. Serum and CSF levels of antigen-bound Aβ-autoantibodies were found to be significantly higher in AD patients compared to age and gender-matched control subjects [201]. It has been suggested that these autoantibodies may well be derived from antibodies raised to pathogens via antigen mimicry. Among the many pathogen proteins showing sequence homology to antigenic regions of Aβ are those from B. burgdorferi, C. Neoformans, C. pneumoniae, H. pylori, P. gingivalis, and herpes viruses (HSV1, HHV6, and the cytomegalovirus) [80, 202]. Cross-reactivity between antibodies to HSV-1 and synuclein (another antimicrobial peptide [160]) has also been experimentally demonstrated [203]. Regardless of the origin of these autoantibodies, their potential to attenuate the antimicrobial effects of Aβ could well contribute to systemic pathogen accumulation, thus rendering cerebral entry more likely. It has already been suggested that Aβ might be used as a therapeutic antibiotic agent [204]. By the same criteria, the removal of blood-borne Aβ autoantibodies by immunoadsorption, to restore such potential, might be considered as a therapeutic option in AD. Autoantibodies to the alpha1-adrenergic receptor may also play a pathological role in relation to the AD vasculature and their immunoadsorption has resulted in a stabilization of cognitive and mental condition in a small group of AD patients [205].
In summary, BBB disruption is promoted by aging and can also be influenced by viral infection via the nasal route, which attains the brain regions first affected in AD. Other bacterial and fungal pathogens also have different ways of circumventing or damaging the BBB. Aging related immunosenescence and autoimmune blockade of the antimicrobial effects of Aβ would contribute to pathogen survival in the periphery. Nasal immunosenescence occurs at a later stage [179], thus limiting the naso-cerebral route to the elderly. The pro-inflammatory gain of function provided by immunosenescence and by certain AD gene variants suggests that cerebral pathogen entry in the aged would be met by a vigorous pro-inflammatory response that likely plays a major role in the degenerative process.
Other environmental risk factors in AD disrupt the BBB and BBB integrity is maintained by beneficial factors
Apart from pathogens, many other environmental risk factors have been reported in AD. These include diabetes, midlife hypertension or obesity, smoking, and physical inactivity [206]. Other contributory factors include previous head injury [207], exposure to toxic metals (aluminum [208, 209] or copper [210]), pesticides (organochlorine and organophosphate insecticides) [211, 212], industrial chemicals (flame retardants) and air pollutants (particulatematter and ozone [213–215]). High levels of cholesterol or homocysteine [216–219] and low levels of folic acid [220, 221] have also been associated with AD. In relation to cholesterol, atherosclerosis of the carotid arteries or of leptomeningeal vessels and in the circle of Willis has also been observed in AD. Such atherosclerotic effects can lead to the chronic cerebral hypoperfusion that is also a feature of AD [127, 223]. Orthostatic hypotension has also been associated with AD, dementia and vascular dementia and this too can lead to cerebral hypoperfusion [224]. Sleep disruption or obstructive sleep apnea are also associated with AD risk[225, 226].
Factors reported to be of benefit, or that reduce the incidence of AD include the use of non-steroidal anti-inflammatories (NSAIDs) or histamine H2 receptor blockers [227–229], and the early use of statins [230–232].
Beneficial dietary factors in AD include caffeine [219], chocolate (versus cognitive decline in the non-demented aged) [233]) and the Mediterranean diet [234–236]. Melatonin [237, 238], estrogen [239–241], and memantine [242] also have reported benefits in AD.
The environmental risk factors associated with AD disrupt the BBB, and BBB integrity is maintained by the beneficial factors (Table 1). While infections are random uncontrollable events, many of the other environmental risk factors are modifiable by lifestyle changes, for example diet, obesity, smoking, and exercise, and it has been estimated that addressing such modifiable risk factors could result in a significant reduction in the incidence of AD [206]. Interestingly, some of these beneficial factors also possess antimicrobial effects, suggesting a multifactorial action. For example, statins have broad spectrum antimicrobial effects (certain oral bacteria as well as viruses and fungi) [243], while NSAIDs have antibacterial effects related to an ability to inhibit bacterial DNA replication [244]. Melatonin also has antimicrobial effects against bacteria and viruses [245] and memantine also has antiviral effects against a neurotropic strain (HCoV-OC43) of a human respiratory coronavirus [246] as well as the influenza or HIV-1 viruses [247, 248].
The effects of Alzheimer’s disease (AD) environmental risk factors and beneficial agents on blood-brain barrier (BBB) function
Amelioration of BBB disruption has already been proposed as a potential therapy in AD, and several drugs including angiotensin receptor blockers, etodolac (NSAID), granisetron (5HT3 serotonin receptor antagonist), or beclomethasone (corticosteroid) [249, 250] as well as other NSAIDS, statins, and other drugs referenced in Table 1 might beconsidered as suitable candidates.
Caveats
This analysis is based on overlapping gene symbols rather than on specific polymorphisms. There is thus no indication of the physiological weight or importance of any gene/pathogen interaction, some of which will be more important than others. Pathogen effects may also be strain-dependent, and the size of the interactomes also varies widely. Within any large interactome there will be deleterious, neutral and beneficial effects. While HSV-1 infection causes Aβ deposition and neurodegeneration [251], in its latent form, the virus can have neuroprotective effects. For example the viral latency transcript inhibits apoptosis and promotes neurite sprouting in neuroblastoma cells [252], protects neuronal C1300 and Neuro2A cells from granzyme B-induced apoptosis and CD8 T-Cell killing [253] and also protects trigeminal neurons from apoptosis [254]. The Bornavirus is capable of promoting hippocampal degeneration in Man [11]. In rats Borna virus infection decreases choline acetyltransferase activity in the cerebral cortex, horizontal diagonal band of Broca, hippocampus and amygdala [255] a situation similar to that observed in AD [256] but the inflammation and microglial activation it produces can also reduce Aβ immunoreactivity in the brain parenchyma of Tg2576 mutant Aβ mice [13]. In adult mice, 12 months after infection, T. Gondii causes neurologic and behavioral abnormalities that are secondary to inflammation and neuronal loss. Activated microglia were also seen in perivascular areas and the brain parenchyma [257]. T. gondii infection in BALB/C mice also inducesneuroinflammation and learning and memory deficits. It also potentiates the toxic effects of low doses of intracerebrally administered Aβ [258], but chronic infection can also increase Aβ phagocytosis and clearance by recruited monocytes [259].
Dementia or neurodegeneration, in the absence of amyloid plaques is, by current clinical definition, not considered as AD, but as already noted, there is no inherent biological reason for this[260, 261]. Such divergent effects might also be relevant to findings relating to the presence of amyloid plaques in the absence of dementia, as observed in the Nun study [262, 263] or to diagnosed AD in the absence of Aβ. A recent report showed that ∼15% of patients clinically diagnosed with AD do not have amyloid deposits as indexed by positron emission tomography [264]. While some amyloid-negative patients could be re-diagnosed (∼50% ), the clinical follow-up using other criteria in other amyloid-negative patients continued to support the definitionof AD.
There are also many inter-pathogen interactions relevant to this relatively small sample of the potential microbiome. For example HSV-1 infection activates replication of the EBV [265]. Gingipains or other proteases secreted by P. gingivalis degrade multiple complement components [266] as well as alpha- and beta defensins [267], immunoglobulins, IgG1 and IgG3 [268], and interleukin-12, preventing its ability to stimulate interferon production [269]. Such effects enable the pathogen to counteract immune defense and would also impinge on the viability of many other pathogens.
HIV-1 is immunosuppressant and has been associated with many opportunistic pathogens including tuberculosis, toxoplasmosis, cytomegalovirus encephalitis and Cryptococcal brain invasion[270, 271]. The human cytomegalovirus is also immunosuppressant via an ability to target MHC class I molecules for degradation [272] and to inhibit MHC class II antigen presentation [273]. Parasites, which maintain a long-term, if unwelcome presence in the host have also developed immunosuppressant and anti-defensive strategies [274, 275]. In addition, the success of most pathogens depends upon their ability to subvert the defensive armory of the host in some way.
The AD genes affect human processes relevant to the disease itself, but given that they are also part of pathogen interactomes, polymorphisms therein are also likely to affect pathogen life cycles or the ability of pathogens to promote diverse effects within the host. Apart from the APOE4 allele there are no studies relating to the effects of the AD gene variants on pathogens or their effects.
For these and many other reasons, it is perhaps unwise to rank the pathogens by order of importance in relation to their enrichment or p value in any of the data described above. Suffice it to say that diverse pathogens have been detected in the AD brain and all of the bioinformatics data presented above, whether related to genes, transcriptomes, plaques or tangles implicate multiple species of pathogens across viral, bacterial, fungal, and protozoan classes.
While there are statistical limitations to this type of analysis, correction for false discovery followed by the Bonferroni correction has been conservatively applied. The relationship of AD to pathogens is supported by experimental observation and by the antimicrobial effects of Aβ. This study also relies on multiple and diverse in silico bioinformatic analyses linking AD GWAS genes, plaques and tangles as well as the hippocampal transcriptome to multiple pathogen interactomes, and the upregulated AD hippocampal genes to multiple infection datasets from diverse pathogen species. Polymicrobial involvement is also supported by the diversity of bacterial, viral and fungal sensors and defenders that are upregulated in the AD brain, blood, or CSF. Each comparison relates to single pathogens but given the diversity of pathogens detected in AD such effects are likely to be cumulative.
CONCLUSIONS
Multiple and diverse pathogens (bacteria, viruses, fungi, and spirochetes) have been detected in the AD brain and many cause neurodegeneration, increase Aβ deposition and tau phosphorylation or are killed/incapacitated by Aβ, an antimicrobial peptide that is part of the innate immune defense system. Representatives of these pathogens target multiple AD GWAS genes, and their interactomes are enriched in genes related to the AD hippocampal transcriptome and to the proteins found in AD plaques and tangles. The upregulated genes of the AD hippocampal transcriptome also correspond to those upregulated by multiple species of viral, bacterial, fungal, and protozoan pathogens or by interferon gamma and TLR ligands.
The AD genes are preferentially localized in the bone marrow and other immunocompetent tissues, and in exosomes that are hijacked by pathogens for intercellular spread. They are also localized in the lateral ventricle and the hippocampus which abuts this area, a prime site of pathogen invasion via the choroid plexus and the blood-CSF barrier.
The AD genes are enriched in global GWAS datasets relating to pathogen diversity, suggesting that some have been selected for pathogen resistance rather that susceptibility. This is supported by the old age of AD patients, indicating survival from the many infections that contribute to mortality in the younger population. APOE4 variants protect against malaria and hepatitis C, and immune/inflammatory gain of function applies to APOE4, CR1, TREM2, and presenilin variants, supporting this contention. Logically, any gene variant increasing the production of the anti-microbial peptide Aβ in response to pathogens might also be considered as beneficial in evolutionary terms. Gene networks involved in innate immunity, inflammation and pathogen defense have been subject to positive selection pressure, although many arms of these networks are cytotoxic (for example, cytokines or free radicals), and evolution has involved a considerable trade-off between diseases/infections prevented by and tissue damage caused by excessive immune activation or inflammatory processes[276, 277]. The same might therefore apply to the toxic effects of Aβ in the brain, particularly if, as in old age, pathogens normally barred from access can infiltrate a defective BBB.
Apart from the APOE4 allele, there is, however, little data examining the effects of other AD gene variants on pathogen life cycles or that relate specifically to individual pathogen responses or resistance to pathogens.
Many AD genes are also localized in the BBB. This should provide an effective shield against manyinfections but it is disrupted by multiple environmental risk factors implicated in AD and protected by several factors reported to be beneficial in relation to AD, including NSAIDs, statins, estrogen, memantine, melatonin, and components of the Mediterranean diet. In addition, viruses such as HSV-1 which can enter the brain via nasal/olfactory pathways are known to attain and damage basal forebrain areas which control the cerebral vasculature and the BBB. This olfactory route relates to the known progression of AD.
The relationship between pathogens and AD has a long history coupled with a degree of skepticism, perhaps related to an inability to fulfil Koch’s postulate. For example, the same pathogen is not always found in all AD brains, or in different laboratories, and independent confirmation is required for several species of pathogen. Laboratory confirmation in animal models may be impossible for certain pathogens, for example the EBV or hepatitis C virus, that do not infect rodents. Nevertheless, the diversity of pathogens able to promote neurodegeneration, Aβ deposition or to mimic the effects observed in the hippocampal AD transcriptome suggests that many candidates, alone or severally, could be involved in the pathogenesis of AD. A polymicrobial involvement seems likely given the multiple species detected in the AD brain. Evidently, this could be assessed by microbiome studies in the periphery or in postmortem brains.
Recent work suggests that the production of the antimicrobial/antiviral peptide Aβ is an expected consequence of infection in general [47, 48]. In the context of the amyloid hypothesis which posits a causal role for Aβ in AD [46], this places pathogens upstream of Aβ, and logically as causal, both in terms of Aβ production and in relation to AD.
Two separate case reports have shown remission from an initial diagnosis of dementia or AD in patients subsequently diagnosed with and treated for C. neoformans infection [278, 279].
In a Greek study, H. pylori-infected AD patients receiving the triple eradication regime (omeprazole, clarithromycin and amoxicillin) showed improved cognitive and functional status parameters where bacterial eradication was successful [280]. H. pylori eradication in AD patients with peptic ulcer was also associated with a decreased risk of AD progression in a Taiwanese study [281]. A Canadian study has shown that antibiotic treatment (doxycycline and rifampin) reduced the rate of cognitive decline (Standardized AD Assessment Scale cognitive subscale) in mild to moderate AD [282]. Although few in number, these are perhaps the only studies with any demonstrated effectiveness in slowing the progression of AD.
Taking all of the above into consideration the combined data suggest that polymicrobial brain invasion, favored by environmentally-induced BBB defects and also by autoantibody blockade of the antimicrobial effects of Aβ in the periphery, may be responsible for AD. This could essentially be mediated via activation of an inflammatory network, and cytotoxic defense mechanisms including the call-up of cerebral Aβ that, as a consequence, causes massive neuronal destruction in a tissue incapable of regeneration. The role of the innate immune system and the inflammatory response in neurotoxicity has recently been reviewed, and innate surveillance mediated cell death has been suggested as a plausible common pathogenic pathway responsible for many neurodegenerative diseases, including AD [283].
It is therefore not unreasonable to suggest that antibiotic, antifungal, and antiviral agents, possibly in combination, tailored to the individual, might be able to halt, delay, or perhaps even provide remission in patients with AD.
CONFLICT OF INTEREST
The author reports no funding and no conflict of interest.
Footnotes
ACKNOWLEDGMENTS
Thanks are due to the many authors who have sent reprints and supplementary datasets that made this work possible. Thanks also to David Eby from the Broad Institute of MIT and Harvard for his help with the Molecular Signatures database.