
Abstract
The aim of the present study was to review the available literature on the evolutionary processes that have led to the development of the human central nervous system (CNS) cholinergic system and to test the hypothesis that such processes may have contributed to the emergence of psychiatric diseases. First, it is clear that the molecular components that have come together to form the cholinergic system in the human CNS initially had functions that were not involved in neurotransmission. Indeed, components of the cholinergic system of the human CNS may be present in nearly all forms of life and may have existed since life began. Significantly, components of the human CNS cholinergic system had begun to interact with each other millions of years ago, long before the appearance of any nervous system. Moreover, there are data to suggest that changes in the mammalian CNS cholinergic system can involve involved gene and environment interactions. Evidence is available to support the notion that the changes in functioning of the human CNS cholinergic system in individuals with psychiatric disorders may have resulted from both Darwinian and Lamarckian concepts of evolution.
As we celebrate the 150th anniversary of the publication of Charles Darwin's theory of evolution [1], it is significant that arguments raging at that time have become relevant to current thinking on the causes of psychiatric disorders. Darwin advocated that evolution was driven by random behavioural within adaptations a species, with some of these random adaptations making an organism better suited to survive in an environment. This better adaption would result in increasing number of progeny and the increasing dominance of the ‘improved’ species within the environment. In today's parlance, it would be argued that evolution is underpinned by the random variations that occur during DNA replication and that some of these random variations produce changes in a species genome that in turn better adapt an organism to survive within an environment or to expand into a new environment. The outcome of this process is that the organisms with the altered genome prosper and the changed gene sequence passes unchanged through subsequent generations [2]. More succinctly, however, Darwin indicated that his concepts equated to the ‘preservation of favoured races in the struggle for life’.
At the time of publication, Darwin's thesis was interpreted as being in conflict with the theory of transmutation as proposed by Lamarck [3]. The underlying premise of that theory was that change in living organisms occurred because they (i) acquired new characteristics as they adapted to their environment; and (ii) were able to pass these new characteristics to their offspring, making the organism more adept at living in the environment. It is now increasingly recognized that environmental factors can act through epigenetic mechanisms, such as changes in levels of gene promoter methylation, to regulate gene expression [4]. Moreover, while epigenetic mechanisms were predicted not to be heritable, trans-generational epigenetic effects have now been identified [5]. This arises the possibility that, through trans-generational epigenetics, the environment can influence the information passed between generations. Thus, it is possible that we are close to verifying the ideas of both Lamarck and Darwin in that change in DNA sequencing and environmentally modulated-heritable changes in gene expression could contribute to how well an organism can adapt to changing environments.
A growing understanding of the interactions between genes and environment has also prompted the suggestion that gene × environment interactions are important in the genesis of psychiatric disease [6]. It has also been argued that adaptations underpinning the development of the human central nervous system (CNS) have also led to the derangements in CNS function that cause psychiatric disorders and that these changes are unique to Homo sapiens [7,8]. Thus, being aware of how evolution has acted to shape CNS function could help in understanding the cause of psychiatric diseases.
There is a large body of evidence that implicates abnormalities in neurotransmitter systems in the pathophysiology of schizophrenia [9], bipolar disorder [10] and major depressive disorders [11]. In considering the impact of evolutionary processes on the development of neurotransmitter systems, it is clear that the components of these systems did not initially form complex pathways designed to transmit information around the CNS. Rather, what are now critical components of neurotransmitter systems had quite different functions when they first evolved [12]. These functions must have been advantageous, allowing the survival of what are now components of the cholinergic system of the human CNS.
This review will focus on current thinking as to how components of the cholinergic system have been influenced by evolution. The focus on the cholinergic system is because it (i) sets boundaries for this review; (ii) acknowledges that acetylcholine was the first chemical entity to be identified as having a role in neurotransmission [13]; and (iii) is relevant to this Journal because the cholinergic system is increasingly thought to be affected by the pathophysiology of psychiatric disease [14,15].
Cholinergic system: molecular overview
This review will consider the evolution of the molecular components of the human CNS cholinergic system, acknowledging that other important aspects of cholinergic neurotransmission have been explored in excellent omnibus reviews [14,16].
Of course the evolution of the molecular components of the CNS cholinergic system cannot be traced through the fossil record, but notions on its evolution can be inferred from the study of the distribution of its different components throughout living organisms and, more recently, the preservations of gene sequencing and functionality between these organisms. As the name suggests, central to the cholinergic system is acetylcholine (Figure 1). Unlike many neurotransmitters, acetylcholine is synthesized at the synapse from acetyl coenzyme A and choline by choline acetyltransferase (Figure 2) [17]. Acetyl coenzyme A is supplied by the metabolism of glucose while choline is taken up from the extracellular milieu by choline transporters, with the intra-cellular concentration of choline being the limiting factor in the synthesis of acetylcholine. As with other chemical neurotransmitters, once synthesized, acetylcholine is taken into synaptic vesicles by a vesicular transporter, the acetylcholine vesicular transporter. Release of acetylcholine involves the fusion of neurotransmitter containing vesicles with the neuronal membrane following a depolarization signal.
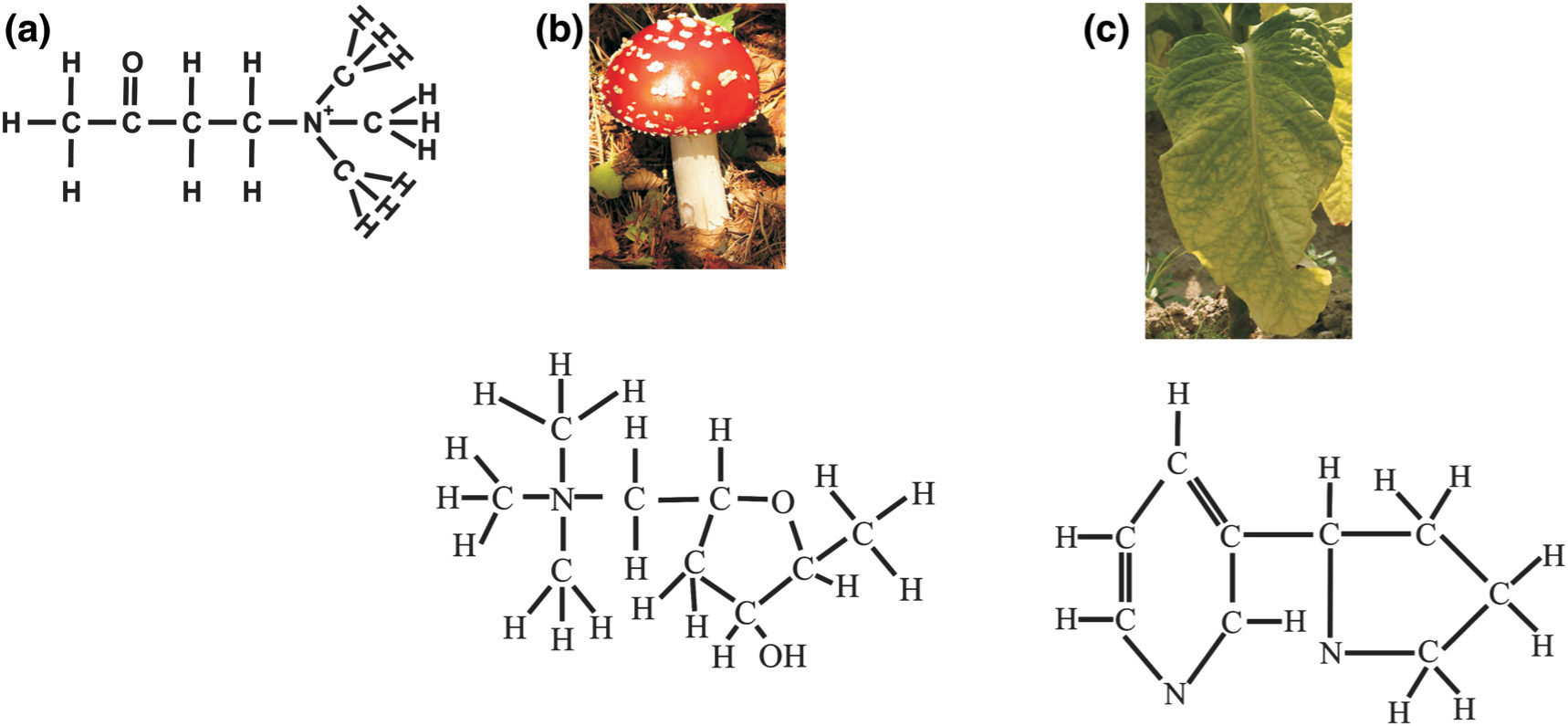
Schematic structures of (a) acetylcholine, (b) muscarine (from Amanita muscaria) and (c) nicotine (from the Solanaceae family).
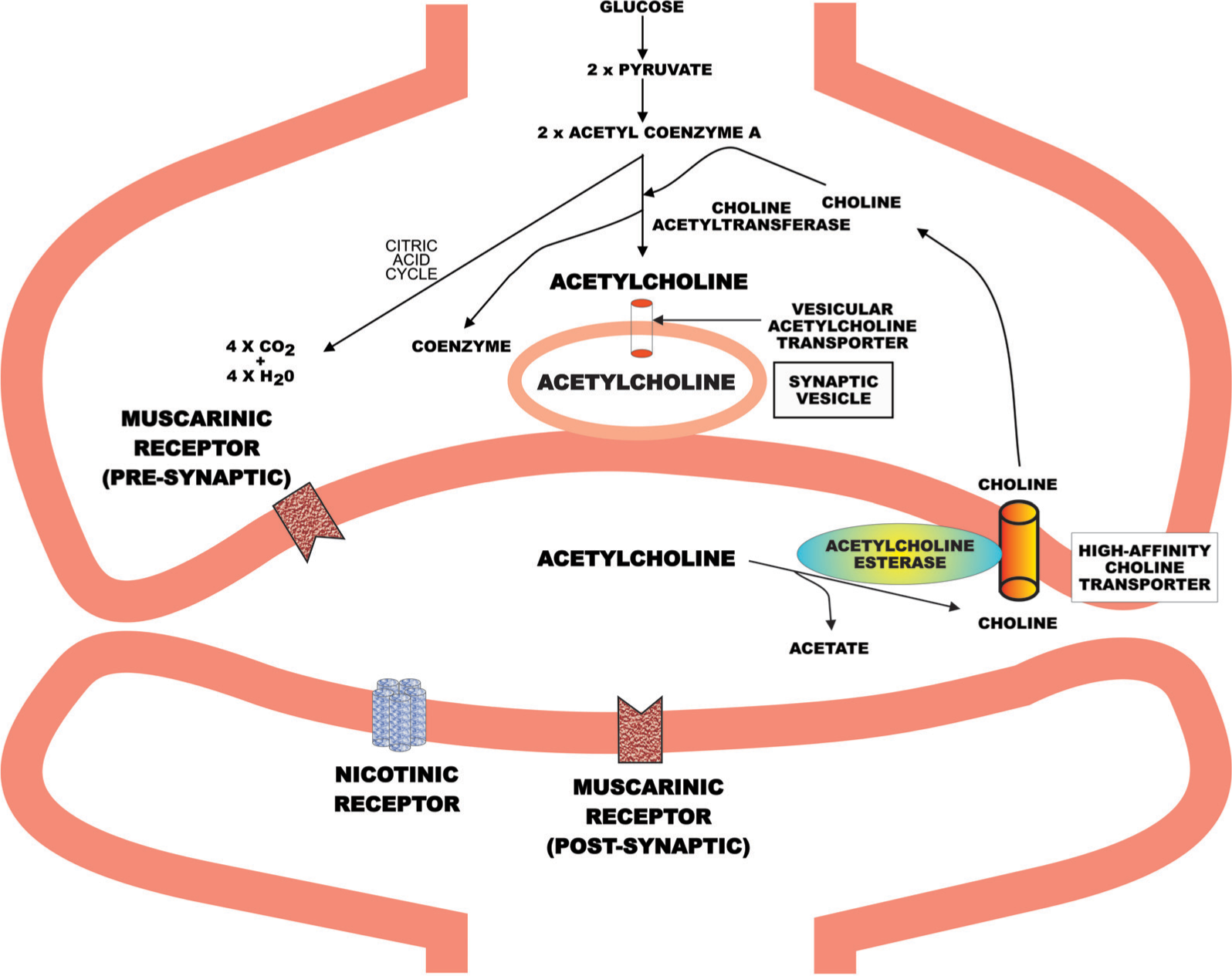
Molecular components of the cholinergic system. Unlike many other neurotransmitters, acetylcholine is synthesized at the synapse rather than in the cell body of the neuron.
Acetylcholine, when released into the synaptic cleft, may be rapidly degraded by acetylcholinesterase to give acetate and choline, with some of the choline being taken up by the choline transporter. Thus, unlike other chemical neurotransmitters there is no acetylcholine transporter on the cellular membrane that removes the transmitter from the synaptic cleft. Acetylcholine that avoids degradation can act on two families of receptors, termed the muscarinic and nicotinic receptors. Muscarinic receptors are so called because they are activated by muscarine, which was first isolated from the mushroom Amanita muscaria (Figure 1); these receptors are G-protein coupled [18]. The other family of receptors are ligand-gated ion channels and are termed nicotinic receptors, because they are activated by nicotine, which is found in the Solanaceae family of plants (e.g. tobacco; Figure 1) [19].
Thus, the human CNS cholinergic system is a complex system at the molecular level and this review will mainly consider what steps in evolution were critical to its creation.
Choline and the choline transporter
In beginning to consider the cholinergic system and its components, it has been known for some time that choline uptake occurs in a wide variety of living organisms. In plants, choline can be metabolized from ethanolamine [20], which means that plants have the capacity to produce sufficient choline for their requirements. By contrast, it has been suggested that mammals can synthesize only approximately 15% of their daily choline requirements, with the balance being obtained as an essential dietary component [21], mainly in the form of lecithin. Following choline been taken up by a cell it can be utilized to synthesize phosphatidylcholine, a membrane phospholipid [22]. Notably, the choline that is converted to phosphatidylcholine does not appear to be available for synthesis of acetylcholine. But the fact that choline is required by most living organisms, ranging from unicellular to Homo sapiens, shows that it (i) has a long evolutionary lineage; and (ii) was present in living organisms before there was a need for a CNS cholinergic system.
Given the limited capacity for synthesis, most organisms need to take up choline from the extracellular milieu, but the need for choline is not universal because choline-derived phospholipids are not common in bacterial membranes [23]. Some bacteria, do require choline and display active choline transport via a choline transporter [24], which, in some bacteria is inducible by the presence of choline. Importantly, most bacterial choline transport seems to involve a system homologous to the rat low-affinity uptake mechanism [25], which provides choline for the synthesis of phosphatidylcholine rather than the synthesis of acetylcholine. This suggests that the initial drive to develop a choline transporter was linked to the need for choline for membrane synthesis rather than the genesis of a neuronal-based cholinergic signalling pathway.
Staying focused on unicellular organisms, it has been shown that in certain bacteria (e.g. Pseudomonas aeruginosa [26] and P. fluorescens [27]) choline can induce the transcription of cholinesterase. This suggests that there is a requirement for unicellular organisms to break down acetylcholine. Notably, certain Pseudomonas such as P. aeruginosa have high- and low-affinity choline uptake mechanisms [28]. The need for two transporters likely reflects the beginning of the duel use of choline for the synthesis of phosphatidylcholine and acetylcholine, and would be the beginning of unicellular organisms needing acetylcholinesterase to control levels of acetylcholine. For this hypothesis to be at least partly proven, it would need to be shown that bacteria having a high-affinity choline transporter always expressed acetylcholinesterase.
Unlike the mammalian neuronal high-affinity cholinergic transporter, the high-affinity transporter in Pseudomonas is not sodium dependent and therefore the two mechanisms are not homologous. It is reasonable, however, to infer that the evolution of the neuronal high-affinity choline transporter has its origins in the high-affinity transporter present in unicellular organisms. Significantly, the neuronal high-affinity choline transporter is readily identifiable in all mammalian CNS [25] and has a highly conserved gene sequence [29]. This suggests that the development of the neuronal high-affinity choline transporter was critical to genesis of cholinergic neurotransmission.
The relationship between choline and the choline transporters has become more complex since the discovery that most of the choline derived from the breakdown of acetylcholine is taken up by the high-affinity choline transporter [30]. This suggests that acetylcholinesterase and the high-affinity choline transporter are in close proximity on the cell membrane (Figure 2). More recently it has become apparent that the extracellular concentration of choline is at levels that would saturate the high-affinity choline transporters that are available at steady state [31]. This was surprising because it meant there would be no capacity to increase choline uptake, which is known to occur after the release of acetylcholine. It is now known that the increase in choline uptake following acetylcholine release is possible because neurotransmitter release triggers a translocation of high-affinity choline transporters from the cytosol into the membrane [31], providing an increased capacity for choline uptake.
Current data would argue that the earliest evolutionary events that provided the foundations for the development of human CNS cholinergic transmission were (i) the appearance of the high-affinity choline transporter; (ii) the ability of acetylcholine release to trigger the translocation of the transporter to the cell membrane, where it can act to increase choline uptake; and (iii) the localization of the high-affinity transporter and acetylcholinesterase in close proximity.
In attempting to date the possible genesis of these events it is noteworthy that a high-affinity choline transporter has been identified in the bacteria Rhizobium meliloti [32], and that there is a high level of conservation of the high-affinity choline transporter gene from Caenorhabditis elegans to Homo sapiens [33]. It is estimated that bacteria were present on earth approximately 4 billion years ago and that Caenorhabditis elegans and Homo sapiens diverged >1 billion years ago [34]. Thus, it would seem likely that many of the critical components of the human CNS cholinergic system have been functional and interactive for at least l billion years.
Acetylcholine: a molecule with diverse functions
Acetylcholine has long been recognized to have functions that pre-date neurotransmission because it is present in organisms that lack a nervous system [35,36]. Indeed, acetylcholine has been isolated from representatives of the three domains of life: the bacteria, the eucarya (plants) and the archaea (animals) [36], including marine animals, eggplant, trees, ferns, mosses, fungi and bacteria [37]. The function of acetylcholine in many of these organisms does not involve neurotransmission; for example in P. fluorescens acetylcholine has been shown to inhibit chemotaxis [38], which suggests an early evolutionary role for acetylcholine in the control of movement or in responding to chemical stimuli. Significantly, the same study showed that epinephrine stimulated chemotaxis. This suggests that a role in controlling movement or response to chemical stimuli was a common physiological role for chemical entities, which are now mainly recognized for their role in neurotransmission.
Acetylcholine has been identified in mosses such as Equisetum robustum [39], which have existed as a family for 400 million years, showing that there have been opportunities for functions of acetylcholine to diversify since the separation of the animal and plant kingdom. It is therefore significant that plants have acetylcholine receptors and that through these receptors acetylcholine is able to regulate water uptake [40], germination, flowering, leaf movement, photomorphogenesis, protoplast swelling and ion permeability [41]. The regulation of many of these functions is thought to be achieved by acetylcholine acting on membrane ion channel permeability, probably via nicotinic receptors that are ligand-gated ion channels. Significantly, despite having completely different physiological effects, the modulation of ion channels by acetylcholine is critical to maintaining normal functioning of the human CNS [19]. This once again suggests that the components of cholinergic neurotransmission have functioned in partnership long before the development of nervous systems but that these partnerships have been maintained in the creation of the neuronal cholinergic system.
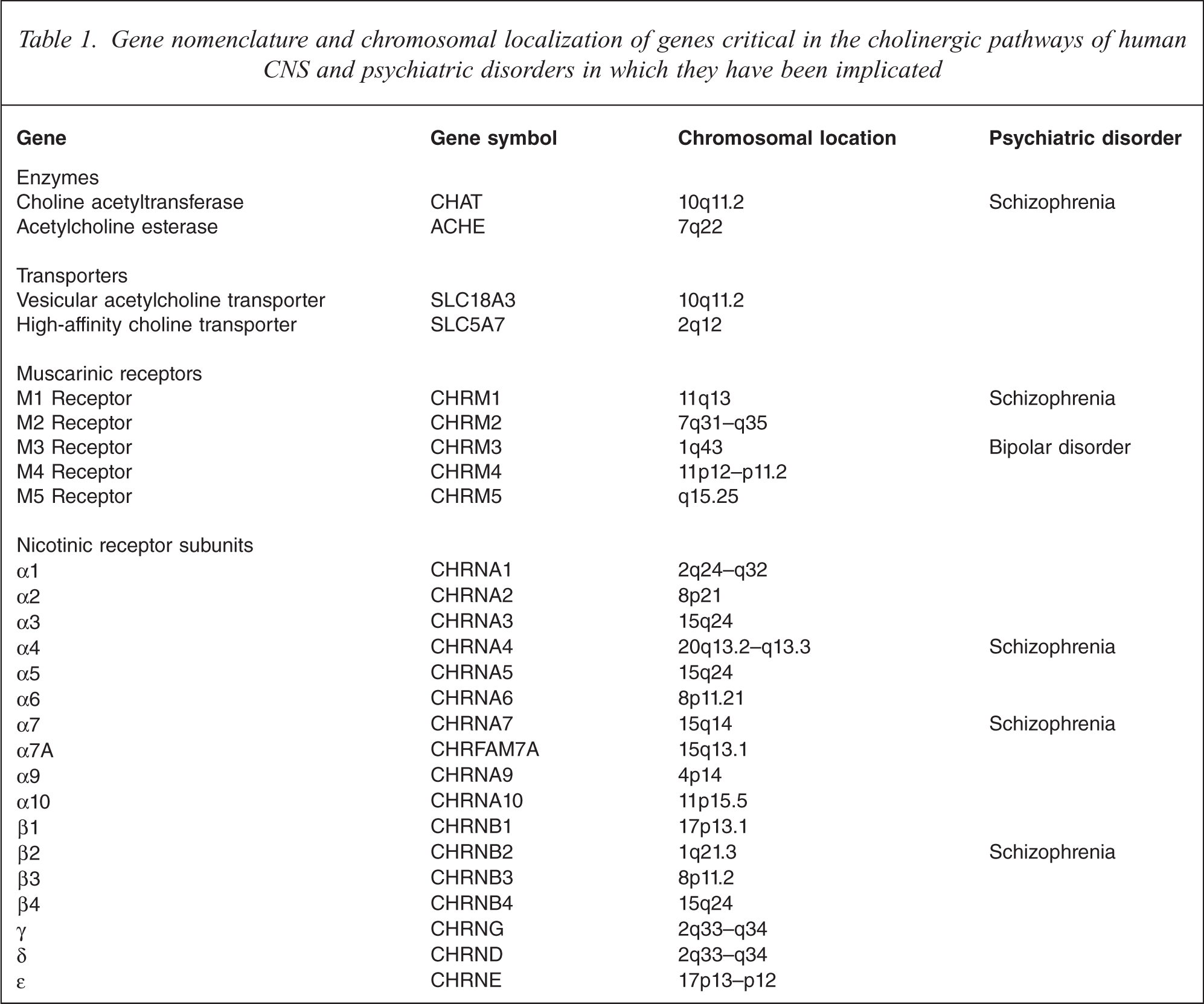
Given the widespread availability of acetylcholine it is necessary to consider what may have been critical in it becoming a significant neurotransmitter in the CNS and periphery. One of the requirements for cholinergic neurotransmission is that acetylcholine can be secreted rapidly in quantal amounts [42]. To allow the secretion of acetylcholine in such a manor the neurotransmitter needs to be packaged into synaptic vesicles; the sequestering of acetylcholine into vesicles is carried out by the acetylcholine vesicular transporter. The acetylcholine vesicular transporter is present in insects [43], invertebrates [44] and fungi [45], which shows that even this critical component of the CNS cholinergic system did not emerge solely to have a role in the cholinergic nervous system. Two interesting observations concerning the vesicular acetylcholine transporter are that it is able to influence the quantal size of neurotransmitter release [42,46], and that the gene encoding the transporter in rat [47] and human [48,49] is within the first intron of the acetylcholinesterase gene. These data suggest that the full interactivity of the components of the human CNS cholinergic system may not yet be fully understood and that there may have been some evolutionary pressure to place and to maintain genes encoding components of the cholinergic system in close proximity (Table 1).
Gene nomenclature and chromosomal localization of genes critical in the cholinergic pathways of human CNS and psychiatric disorders in which they have been implicated
One interesting development in humans relating to the cholinergic system is that choline is not available to the fetus from maternal blood because the placenta does not supply choline to the fetus [21]. Rather, choline from maternal blood is taken up by the placenta by a passive and an active uptake mechanism [50], after which it is mostly rapidly converted to acetylcholine [50]. It would then appear that acetylcholine passes from the placenta to the fetus, where it is hydrolysed to give a source of choline. The notion that this process is related to fetal development is supported by the finding that the level of acetylcholine produced by the placenta is a controlled process, with the highest level of acetylcholine being reached between 16 and 24 weeks gestation [51]. It is still not clear why such a complex system has arisen, and hence shows that much is still to be learned about the role of cholinergic systems, not the least of which might be whether derangements in the supply of acetylcholine to the fetus could affect its development.
Molecular clock: genetic evolutionary dating system
Initially, the effects of evolution on different molecular systems relied upon knowing the distribution of particular markers, such as acetylcholine, across living organisms. This changed significantly with the advent of cloning technologies, which have allowed theories to be constructed about the evolution of families of proteins based on gene sequence conservation and divergence. The evolutionary change or selection pressure on proteins can be measured using either the ratio of the rate of non-synonymous to synonymous nucleotide sequence substitution [52] or the ratio of radical to conservative amino-acid replacement rates [53]. In the case of the first approach, non-synonymous changes in the DNA sequence change the amino-acid sequence of a protein and therefore must be in a gene exon. By contrast, non-synonymous changes in the DNA sequence do not change the amino-acid sequence of a protein and can therefore be in the coding or non-coding sequences of the genome. Importantly, a synonymous/non-synonymous substitution rate >1.0 is taken to indicate a positive selection during evolution [54]. With regard to conservative amino-acid replacement rate it is postulated that amino-acid substitutions that involve a change in charged amino-acids reflects positive evolutionary pressure [53]. The second approach is useful in assessing evolutionary pressure in distantly related coding sequences because, in such cases, the synonymous substitution rate [54] can become saturated. For the present review it is sufficient to be aware that these processes have allowed estimates of evolutionary divergence to be made by comparing gene sequences within and between species.
Acetylcholine receptors: evolutionary adaptations to diverse roles
One of the earliest advances in understanding of the mechanisms underlying chemical neurotransmission was the identification of two classes of receptors for acetylcholine [13]. As stated earlier, the two classes of receptors were first separated based on the binding profiles of two agents, muscarine and nicotine, both of which were derived from plants [55,56]. This highlights the preservation of the ligand-binding domains of the cholinergic receptors in the face of significant changes in the gene sequences that have occurred since the divergence of plants and animals. Being contentious, it could be argued that the preservation of binding sites for muscarine and nicotine might indicate that the role of cholinergic receptors is not solely devoted to acetylcholine signalling, but that it also allows signalling by entities such as muscarine and nicotine. Alternatively, it could be argued that there has been no great evolutionary pressure for the receptors to discard the ability to bind ligands other than acetylcholine. This conundrum illustrates the problems in trying to interpret the biological reason for a gene to conserve a function.
Cloning technologies have allowed the complexities of the cholinergic receptors to be unravelled. Thus, the family of muscarinic receptors is now known to be made up of five gene products with the nomenclature of muscarinic M1, M2, M3, M4 and M5 receptors (Table 1) [57]. By contrast, nicotinic receptors are made up of distinct proteins that come together as pentomers to form a ligand-gated ion channel (Figure 3) [19]. These proteins, which are now commonly known as nicotinic receptor subunits, are individual gene products (Table 1) and have been divided into families according to sequence and structural homologies. In the case of α-subunits this means that all the subunits have a cysteine–cysteine pair near the entrance of the first transmembrane domain. In neuronal tissue nine genes have been cloned and designated as-subunits (α1, α2, α3, α4, α5, α6, α7, α9 and α10: note α8 was found in birds but has yet to be found in mammals), while four other genes have been designated as β-subunits (β1, β2, β3 and β4; Table 1; Figure 3). The complexity of the nicotinic receptor structure is perhaps one of the most interesting outcomes of the evolutionary process and this complexity appears to be a critical factor in the functioning of the human CNS.
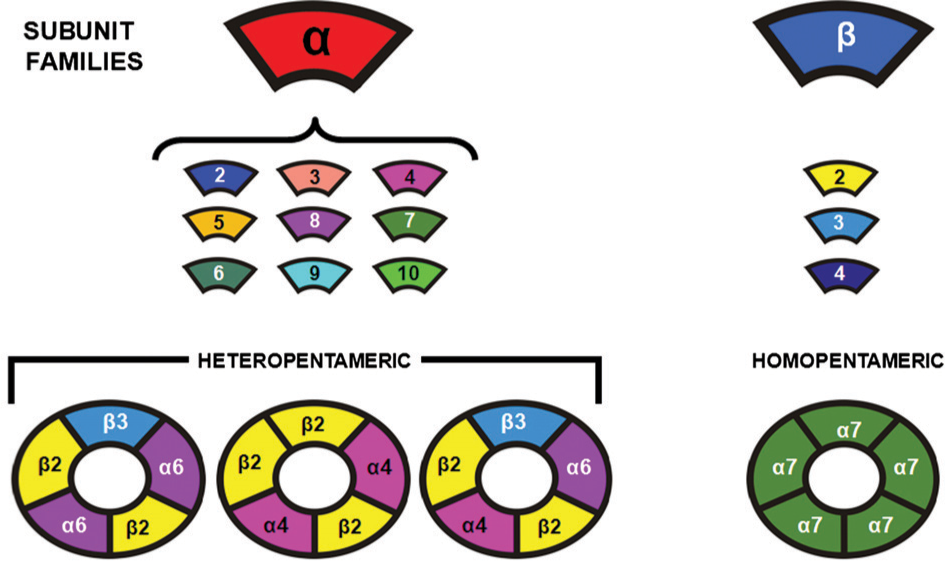
Schematic showing how nicotinic receptor subunits come together to form homo- and hetero-pentomeric ligand-gated ion-channels
Initially it was suggested that cholinergic receptors were not present in the kingdoms of Protoctista and Fungi or in the phyla Porifera and Cnidaria [58]. It has subsequently been shown that cholinergic receptors are present in Paramecium primaurelia, in which they appear to have a role in controlling mating pair formation. A review of current databases show that gene sequences with similarities to the muscarinic M2 and M3 receptors have been predicted from study of the sea urchin genome (Strongylocentrotus purpuratus), and that genes encoding muscarinic receptors have been identified in Drosophila melanogaster [43], mosquitoes (Anopheles gambiae) [59] and Caenorhabditis elegans [60]. Surprisingly, there are no listed sequences for nicotinic receptors in bacteria, fungi, plants or invertebrates. There is a sequence with homology to nicotinic receptor subunits in the zebra fish Danio rerio [61]. Given the strong pharmacological evidence for nicotinic receptors in lower organisms [58], it would seem likely that the absence of data is due to the lack of comprehensive genome screening in many organisms.
In insects there is a growing body of evidence that suggests that the cholinergic system plays a primary role in neurodevelopment and behaviour. Thus, in the moth Manduca sexta nicotinic receptors appear to be involved in controlling the migration of neuroglia in the olfactory system [62], while in the honey bee nicotinic receptors have been shown to be involved in habituation [63]. Also in honey bees, muscarinic receptors are involved in the enlargement of mushroom bodies in the CNS; mushroom bodies being important in honey bee learning and memory [64]. It is significant that muscarinic receptors are important in learning and memory in Homo sapiens [65], suggesting that, despite a high degree of genetic divergence, muscarinic receptors serve similar roles in species that have had a differing genetic lineage for at least 350 million years. It is also significant that abnormalities in learning and memory [66–68] as well as habituation [69–71] have been reported in schizophrenia, bipolar disorder and major depressive disorders, and cholinergic receptors regulate these function in humans. It would therefore appear that abnormalities in behaviours that are observed in individuals with psychiatric disorders have been under the control of the cholinergic system for eons.
The actual time frame over which cholinergic receptors may have evolved has been a topic of much debate. In the case of muscarinic receptors this debate has centred on the changes in gene sequence for the G-protein couple receptors that have occurred since the appearance of the primordial receptor, the rhodopsin receptor [72]. From the study of sequence variation in many genes it is now estimated that the rate of change of non-synonymous sequence substitutions is 0.51 × 10−9 site−1 year−1. Based on this observation, the muscarinic receptor gene family must have separated from the other G-protein receptor super-family of receptors between 0.6 and 1 billion years ago, and the different forms of muscarinic receptors have evolved since that divergence (Figure 4). The nicotinic family of receptors has an equally impressive lineage because it is suggested that this family of receptors evolved from a common ancestor that has been present for at least 2.5 billion years [73]. The first evolutionary divergence in this family of receptors was the separation of the cationic (nicotinic and serotonergic) and anionic (GABA) receptors. A later divergence in the cationic lineage resulted in the separation of the serotonergic (serotonin 3 receptor) from the nicotinic lineage; the family of nicotinic receptor subunits evolving since that divergence.
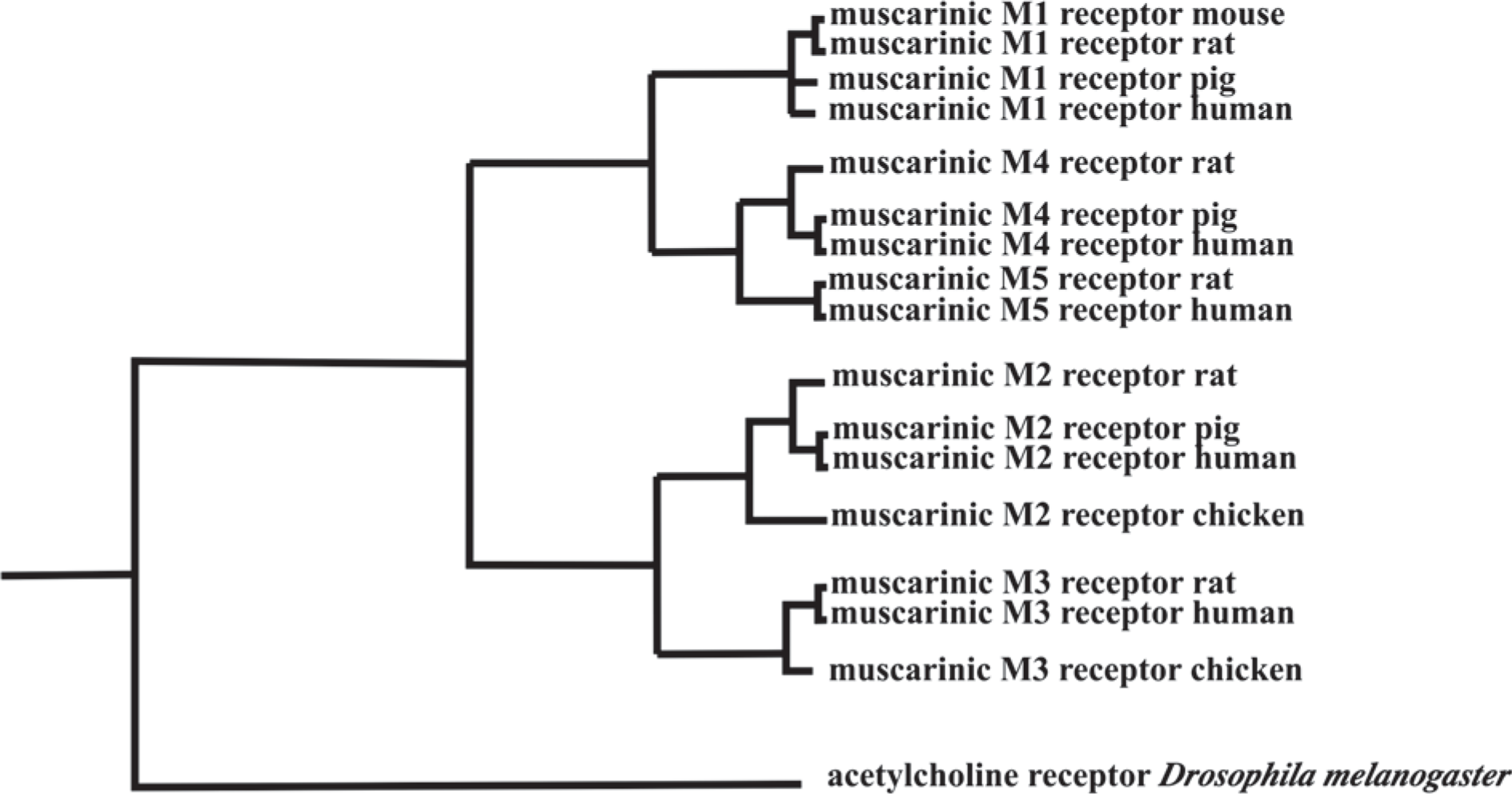
Proposed phylogenic tree for the five muscarinic receptors present in human and rats central nervous system (CNS). Significantly, these pathways show that the muscarinic M1, M3 and M5 receptors and the muscarinic M2 and M4 receptors, which use different signalling pathways [18], resulted from a secondary divergence after the separation of the muscarinic receptors from other G-protein linked receptors.
The first clearly divergent group of nicotinic receptor subunits were the α7 and α8 subunits, which are unique in that they both form homopentomeric receptors [73,74]. The next divergence saw the receptors present in invertebrates separate from those in vertebrates, then the final differentiation of the multiple forms of α, β, δ, ∊ and γ subunits occurred in vertebrates. Significantly, nicotinic receptors exist in muscle and nerves and the evolutionary process leading to this separation is still a matter of debate. On the one hand, it has been suggested that the α1 subunit in muscle evolved from the neuronal α1 subunit [74]. This is because the neuronal α1, α2, α6 and β3 subunits have homologies that suggest they appeared before the muscle α1 subunit. The other argument is that there was a separation in tissue function between 800 and 1400 million years ago and that the differences in neuronal and muscular forms of nicotinic receptor subunits have evolved since that time [73]. Another hypothesis is that evolution resulted in nicotinic receptors subunits forming four types of nicotinic receptors: type I (those that make up the neuronal α-bungarotoxin binding site receptors); type II (the arthropoda neuronal subunit family); type III (the non-bungarotoxin binding vertebrate nicotinic receptor subunit family); and type IV (the muscle subunit family) [75]. This latter proposal essentially agrees with the suggestion that neuronal and muscle tissue diverged prior to the evolution of the separate neuronal and muscle-specific nicotinic subunit families. Whichever of these arguments is correct, this evolution process has produced a range of receptors that have many and diverse functions, a review of which goes beyond the remit of this discussion but which is well worth understanding [19]. Moreover, the role of the cholinergic system in human evolution must be ongoing because it has been suggested that the α2 and α5 subunits are one of a very limited number of genes that appear to be evolving faster in primates compared to rodents [76].
One final twist in the evolutionary story of acetylcholine receptors is that since the divergence of animal and plants, evolution appears to have created a chimeric nicotinic receptor that responds to muscarinic receptor agonists [40]. This receptor has been identified in insects [77], cows [77] and humans [78], and is now recognized as being formed by α9 nicotinic receptor subunits. This subunit is very ancient and may have diverged even before the α7 and α8 subunits [74]. Significantly, in mammals this receptor seems to have become important in systems involved in the transmission of primary sensory information [79,80].
Like muscarinic receptors, CNS nicotinic receptors are critical in controlling CNS functions that seem to be affected in subjects with psychiatric disorders [15,81]. Therefore at this point it would seem appropriate to discuss which components of the molecular cholinergic system are thought to be involved in the pathophysiology of various psychiatric disorders.
Cholinergic abnormalities and psychiatric disease
Early studies on the cholinergic system in the CNS of subjects with psychiatric disease focused on the enzymes within that system. Thus, it was reported that there were widespread increases in the activity of choline acetyltransferase, but not acetylcholine esterase, tyrosine hydroxylase, dopa decarboxylase or glutamic decarboxylase, in the CNS from subjects with schizophrenia [82]. This finding was not confirmed in a later study that reported that the activity of choline acetyltransferase and acetylcholinesterase was not altered in six cortical regions from subjects with schizophrenia [83]. To add to this discrepancy, that study followed a report on a 46% decrease in the levels of the choline acetyltransferase protein in the pontine nucleus, but not frontal cortex, superior temporal gyrus, occipital cortex, thalamus or cerebellum, from subjects with schizophrenia [84]. Finally, decreases in choline acetyltransferase-positive neurons have been reported in the striatum from subjects with schizophrenia [85]. Thus, at present it is difficult to determine if enzymes in the cholinergic system are affected by the pathophysiology of schizophrenia.
Changes in muscarinic receptors in the CNS of subjects with schizophrenia were first suggested by an increase in [3H]quinuclidinyl benzilate ([3H]QNB) binding to orbito-frontal but not medial frontal cortex from subjects with schizophrenia [86]. [3H]QNB binds to all muscarinic receptors and therefore that study could not identify the individual muscarinic receptors that were altered. The next study supporting the notion of changed muscarinic receptors in schizophrenia reported decreased [3H]pirenzepine binding to caudate-putamen from subjects with the disorder [87]. Importantly, due to the method used [88], [3H]pirenzepine binding in that study was a surrogate measure of muscarinic M1 receptors. This becomes significant given later data showing no change in mRNA for muscarinic M1 receptor the caudate-putamen from subjects with schizophrenia [89], suggesting that the change in receptor density was not due to a change in gene expression. In addition, decreased [3H]N-[2-[2-[(Dipropylamino)methyl]-1-piperidinyl]ethyl]-5,6-dihydro-6-oxo-11H-pyrido[2,3-b]1,4benzodiazepine-11-carboxamide ([3H]AF-DX 384) binding to the muscarinic M2 and M4 receptors has been reported in the caudate-putamen from subjects with schizophrenia [90], indicating that changes in receptors other than the muscarinic M1 receptor may be present in that region.
Subsequent to the findings in the caudate-putamen, decreased [3H]pirenzepine binding has been reported in the hippocampus [91], frontal cortex [92], anterior cingulate cortex [93], superior temporal gyrus [94] and posterior cingulate cortex [95], but not the thalamus [96], from subjects with schizophrenia. These data suggest that there are widespread changes in muscarinic receptors in the CNS of subjects with schizophrenia. More recent studies have used more specific tools such as western blotting and quantification of levels of mRNA to better clarify which muscarinic receptors are altered in the CNS from subjects with schizophrenia. These data show that muscarinic M1 receptor protein [97] and mRNA are decreased in the cortex from subjects with schizophrenia. In contrast, there are no changes in levels of protein or mRNA for the muscarinic M2, 3 and 4 receptor or the capacity of muscarinic M 2/4 receptor to bind G-protein in the cortex of subjects with the disorder [97,99]. From these data it would appear that the cortical muscarinic M1 receptor is predominantly affected by the pathophysiology of schizophrenia, a conclusion supported by the finding that [3H]AF-DX 384 binding is not altered in the cortex from subjects with the disorder [94,95]. Significantly, it has been shown that the level of muscarinic M4, but not muscarinic M1, receptor mRNA is decreased in the hippocampus from subjects with schizophrenia [100]. Thus, different muscarinic receptors may be altered in different CNS regions from subjects with schizophrenia.
Changes in levels of muscarinic receptors initially appeared to be limited to schizophrenia, because [3H]pirenzepine binding was not altered in anterior cingulate from subjects with bipolar and depression [93]. A more recent study has confirmed that [3H]pirenzepine binding is not altered in the cortex of subjects with bipolar disorder and depression [15]. The same study, however, found a decrease in [3H]AF-DX 384 binding to muscarinic M2 ++ M4 receptors in the dorsolateral prefrontal cortex from subjects with depression and bipolar disorder, while [3H]1,1-dimethyl-4-diphenylacetoxypiperidinium iodide ([3H]4DAMP) binding to the muscarinic M3 receptor was decreased in the frontal pole of subjects with bipolar disorder. These findings suggest that changes in muscarinic receptors may be involved in the pathophysiology of all of the major psychiatric diseases.
The first indication that nicotinic receptors were affected by the pathology of schizophrenia was the report of decreased [125I]α-bungarotoxin to the hippocampus from subjects with the disorder [101]. Decreased binding of that radioligand has subsequently been reported in the thalamus [102] and cortex [103] from subjects with schizophrenia. Because α-bungarotoxin predominantly binds to α7 subunits of the nicotinic receptor, these data suggested a downregulation of α7 subunit-containing receptors in schizophrenia, a hypothesis supported by the finding that α7 subunit mRNA and protein are decreased in the CNS from subjects with the disorder [104,105]. In contrast, another study has shown that mRNA for the α7 subunit mRNA is not altered in the cortex from subjects with schizophrenia [106].
[3H]nicotine and [3H]cytosine bind to nicotinic receptors that contain α4β2 subunits [107]. Therefore reports of increased binding of these radioligands in the striatum [108], hippocampus [109] and cortex [103] suggest an increase in α4β2 subunit containing receptors in subjects with schizophrenia. Importantly, it is known that smoking significantly increases levels of α4β2 containing nicotinic receptors [110], and levels of smoking are markedly elevated in schizophrenia [111]. Thus, it remains possible that the increase in α4β2 subunits containing nicotinic receptors could be related to differential rates of smoking.
At present it appears that muscarinic and nicotinic receptors are molecular components of the cholinergic system that are most affected by the pathophysiology of schizophrenia.
Concluding remarks
It would appear that the molecular components of the cholinergic system have evolved very early after life began. However, there would seem to have been four critical events that underpinned the development of the neuronal cholinergic system. These were (i) the ability to localize acetylcholine esterase and the high-affinity choline transporter in close proximity; (ii) the development of cells that contained vesicles that could store acetylcholine and a vesicular acetylcholine transporter to load these vesicles with neurotransmitter; (iii) the ability, by those same cells, to regulate the release of acetylcholine following a stimulus; and (iv) the possession, by the cells that release acetylcholine, of, or being in close proximity to other cells that have, acetylcholine receptors.
Clearly these four events come together at the synapse between neurons and at the neuromuscular junction.
Although there is strong evidence to show that the CNS cholinergic system is affected by the pathophysiology of schizophrenia, it is interesting to note that it has been suggested that subjects with acute psychoses display muscular abnormalities [112]. Thus, it remains possible that both the CNS and muscular cholinergic systems have been impacted upon by the pathophysiology of the disorder.
The evolutionary processes that have resulted in the development of the CNS cholinergic system have clearly developed over many millions, if not billions, of years. In fact it has been suggested that components of the system have existed since the beginning of life on Earth [58]. But despite much evolutionary branching of components of the cholinergic systems it seems that many of the behaviours controlled by these receptors have been preserved despite significant gene sequence divergence. There is a growing perception, however, that certain psychiatric disorders, such as schizophrenia, are unique to Homo sapiens [8]. If that proves to be the case then it would be likely that changes in cholinergic function that have been involved in the emergence of the disorder must be of a recent evolutionary origin. Thus, it may be that what has been a very stable biological system has been deranged and has contributed to the emergence of psychiatric disorders? A much better understanding of the evolution of the cholinergic system, the impact of the pathophysiology of psychiatric diseases on that system and the role of that system in CNS function will be required to fully answer that critical question.
As we increasingly understand the complexity of genome divergence, we are now beginning to understand how epigenetic mechanism can be affected by the environment to cause changes in gene expression [5]. But we as yet have no information to determine if these processes could have contributed to the destabilization of the human CNS cholinergic system in subjects with psychiatric disease. It is therefore intriguing that it has been recently shown that single exon genes seem to be overrepresented in genes that have changed levels of expression after an environmental insult [113], because the muscarinic M1 receptor gene is a single exon gene [114]. This raises the possibility that changes in the level of expression of the muscarinic M1 receptor in the cortex of subjects with schizophrenia [14] may be, in part, environmentally driven. This notion is supported by the demonstration that levels of muscarinic M1 receptors in the mammalian CNS can be affected by genotype and changing environment [115]. These latter data open up the possibility that changes in the cholinergic system in subjects with psychiatric disorders may be an outcome of gene and environment interactions. If that is the case, given the possibility that the changes in gene expression that cause schizophrenia may be unique to humans [8], the challenge to understanding the pathophysiology of schizophrenia may involve identifying differences in how epigenetic mechanism can affect gene expression in Homo sapiens and the primates.
In the context of this review, it would seem reasonable to postulate that the cause of psychiatric diseases (including the changes in the cholinergic system) will most likely be associated with some level of genetic sequence variation [116], but will also involve complex gene–environment interactions [6]. In which case, ironically, these disorders may have arisen because of a fusion of outcomes from both Darwinian and Larmarkian evolution.
Footnotes
Acknowledgements
Brian Dean is a Senior NHMRC Research Fellow (Level B) (400016). This research was funded in part by NHMRC Project Grants 192399 and 509333, Rebecca L. Cooper Medical Research Foundation and the Operational Infrastructure Support (OIS) from the Victorian State Government.