
Abstract
Large-conductance calcium- and voltage-dependent potassium (BK) channels are ubiquitously expressed in mammalian cells and participate in various physiological and pathological processes such as neurotransmission and cerebral ischemia. BK channels comprise up to four pore-forming α subunits and zero to four accessory subunits. Although microglial BK currents were initially recorded 27 years ago, their roles have long been elusive. Studies have demonstrated that BK channels modulate the activation, phagocytosis, and probably migration of microglia and have associated microglial BK channels with many neurological diseases, including neuropathic pain and stroke. This review summarizes the available information regarding the biophysical, functional, and pathological aspects of microglial BK channels and discusses future directions of research into these channels.
1 Introduction
Large-conductance calcium- and voltage-dependent potassium (BK) channels are composed of pore-forming α subunits (Slo1, KCNMA1) and auxiliary subunits, including β1 through β4 (KCNMB1 through KCNMB4), γ1 through γ4 (LRRC26, LRRC52, LRRC55, LRRC38), and LINGO1 through LINGO4 [1–7]. BK channels are ubiquitously distributed in the body and play vital roles in various physiological and pathological processes [8, 9]. In the brain, cells including neurons, astrocytes, and microglia all express BK channels on both their plasma membranes and intracellular organelle membranes [10–13]. Although the roles of BK channels in neurons, where they regulate neuronal excitability and neurotransmission [14–16], have been extensively investigated, their roles and functions in microglia have been explored only in a few studies.
Microglia are the immune cells residing in the brain [17]. These cells express a variety of ion channels, including those transporting potassium, calcium, protons, and chloride [18–20], which are involved in the maintenance of resting membrane potential, chemotaxis, phagocytosis, activation, production of reactive oxygen species, and release of cytokines [21–25]. The aim of this review is to summarize the biophysical, functional, and pathological information on microglial BK channels and discuss the future directions concerning these channels.
2 Biophysical properties of BK channels in microglia
In 1995, McLarnon et al. first reported BK-type calcium-dependent K+ currents in cultured primary bovine microglia [26]. By using inside–out excised patches, a large unitary conductance of 240 pS was recorded with symmetrical 140 mM K+ across patches. In their experiments, maintaining 140 mM K+ in the pipette solution while changing the internal K+ concentration strongly affected the unitary currents and channel conductance. At 5, 40, 70, or 140 mM internal K+, the unitary current amplitudes at 0 mV were 4.7, 2.7, 1.6, or 0 pA, respectively. With 5 mM K+ in the internal solution, the unitary conductance was 190 pS. The open probability (P O) of the channel was strongly calcium-dependent (0.9 at 0.2 mM Ca2+ compared with 0.1 at 1 μM Ca2+, −20 mV, and symmetrical 140 mM K+). The voltage for half-maximal activation (V 1/2) was −24 ± 4 or −46 ± 5 mV with 5 or 140 mM K+ in the internal solution, respectively. Macroscopic BK currents were also recorded in 7 of 15 whole-cell experiments, which demonstrated slow activation and a V 1/2 of +21 ± 5 mV. These macroscopic BK currents, which resemble the BK currents in the nerve [27] and muscle [28], were completely abolished when 2 mM tetraethylammonium (TEA) was applied externally.
After 2 years, McLarnon et al. identified BK currents in excised inside–out patches from human microglia isolated from fetal human brains (12–20 weeks of gestation) cultured for at least 5 d [29]. Compared with BK channels from bovine microglia, these channels exhibited a slightly lower unitary conductance (106 pS with internal 140 mM K+ and external 5 mM K+; 205 pS with symmetrical 140 mM K+). However, human microglial BK channels exhibited similar calcium and voltage dependence as bovine microglial BK channels. At 0 mV, reaching a P O of 0.5 required 7 μM Ca2+ in the bath solution with physiological levels of K+ across the patch (internal 140 mM K+ and external 5 mM K+). Further, the V 1/2 was +60 mV with internal Ca2+ at 1 μM. McLarnon et al. recorded currents without differentiating the morphology of the microglia and detected BK currents only in excised patches, whereas Bordey and Spencer specifically focused on amoeboid-shaped microglial cells to record macroscopic BK currents using a whole-cell configuration from microglia in hippocampal slices taken from patients with epilepsy [30]. These channels were activated at 0 mV in nonbuffered internal Ca2+ (0 mM Ca2+/0 mM EGTA) and at +40 mV in 20 nM internal Ca2+. The unitary conductance was 187 pS in cell-attached patches (symmetrical 140 mM K+ across the patches) and 149 pS in outside–out patches (140 mM internal K+/2.5 mM external K+ across the patches). Interestingly, these BK currents were increased 3.3-fold at +60 mV by chemokine macrophage inflammatory protein 1-α (MIP1-α). With limited information, it is difficult to state the most important reasons for the discrepancy between these two studies. The possible factors influencing these differences include the age of the individuals sampled (fetal or adult), the specific pathological event (normal reference or epilepsy), and the functional state of the microglia (resting or activating).
In another two studies, the expression and properties of microglial BK currents in murine brain slices were compared among mice of different ages and among microglia at different functional states [31, 32]. For juvenile C57BL6 mice 5–9 days postnatal, BK currents were only detected in microglia from cultured hippocampal slices in which microglial cells appeared to be in activated states. These BK channels had a single-channel conductance of 164 pS (120 mM internal K+/3 mM external K+) and could be activated at +20 mV with 1 μM Ca2+ contained in the pipette solution [31]. However, for adult (2–3 months) and aged (19–24 months) C57BL6 mice, the microglial BK currents appeared to show no correlation with the functional states of the cell, and there were no significant differences between these two groups in BK current densities recorded from microglia [32]. Notably, the above-described results only describe plasma membrane BK currents. Nevertheless, we found that BK channels are also expressed in the microglial nuclear membrane using western blot and immunocytochemistry methods [12]. Although whether nuclear BK currents resemble plasma membrane BK currents and how they might change with age and the functional state of microglia remain unknown, further investigations on these two questions will shed more light on the functional roles of BK channels in microglia. In addition, Hayashi et al. found that KCNMB3, the β3 auxiliary subunit of BK channels, is exclusively expressed in spinal microglial cells [33, 34]. Whether this subunit exists on both plasma membrane and nuclear membrane and how it influences the biophysical properties of the channel need further investigation. The biophysical properties of microglial BK currents in different samples are summarized in Table 1.
3 Roles of BK channels in microglia
In the juvenile mice hippocampus, BK currents were only observed in activated microglial cells [31]. At 3 d after nerve injury, both the current and protein expression levels of BK channels in spinal microglia were elevated. On the other hand, at 3 d after intrathecal injection (i.t.) with 1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl) phenyl]-5-(trifluoromethyl)-2H-benzimidazol-2-one (NS1619), a specific BK channel opener, the number of spinal microglia increased and the morphology of the cells changed markedly–– cells became larger with shorter processes [35]. In lamina I microglia of mice after 5 d treatment with morphine, the BK currents were increased upon the activation of microglia [33]. These results indicate a close relationship between BK channels and the activation of microglia.
Inspired by results regarding the regulation of BK channels by lipopolysaccharide (LPS) in macrophages [36–40], we used LPS-stimulated murine BV-2 and primary microglial cells as a model to explore the possible roles and underlying mechanisms of BK channels in the activation of microglia [12]. The LPS-induced activation of microglial cells was significantly inhibited by either the blockade of BK channels or knockdown of their expression, confirming that BK channels are involved in the LPS-induced activation of microglia. LPS enhances plasma membrane BK currents through Toll-like receptor 4 (TLR4) in BV-2 cells and then activates BK channels, facilitating the translocation of nuclear factor κB (NF-κB) to the nucleus. Because these events occurred within 30 min after LPS treatment, plasma membrane BK channels probably participate in the very early stages of the LPS-induced activation of microglia. On the other hand, during the first 6 h of incubation with LPS, after the translocation of NF-κB to the nucleus, blocking of BK channels could still suppress the production of nitric oxide, although to a lesser extent. This result suggests the presence of another NF-κB-independent pathway that is most likely associated with nuclear membrane BK channels. The functions of nuclear membrane BK channels in the activation of microglia are still unclear, regulating gene expression (such as the proinflammatory cytokines tumor necrosis factor-α [TNF-α] and interleukin-6 [IL-6]) might be involved, as in the cerebral endothelial cells of Yorkshire piglets [41] and the hippocampal neurons of rodents [42]. Furthermore, the expression of nuclear BK channels is prominently increased by LPS through TLR4 after 12 h of incubation with LPS. Whether this phenomenon is a consequence of the activation of microglia or whether this upregulation plays some roles during the activation of microglia requires further exploration.
Biophysics of microglial BK channels in different species and samples.
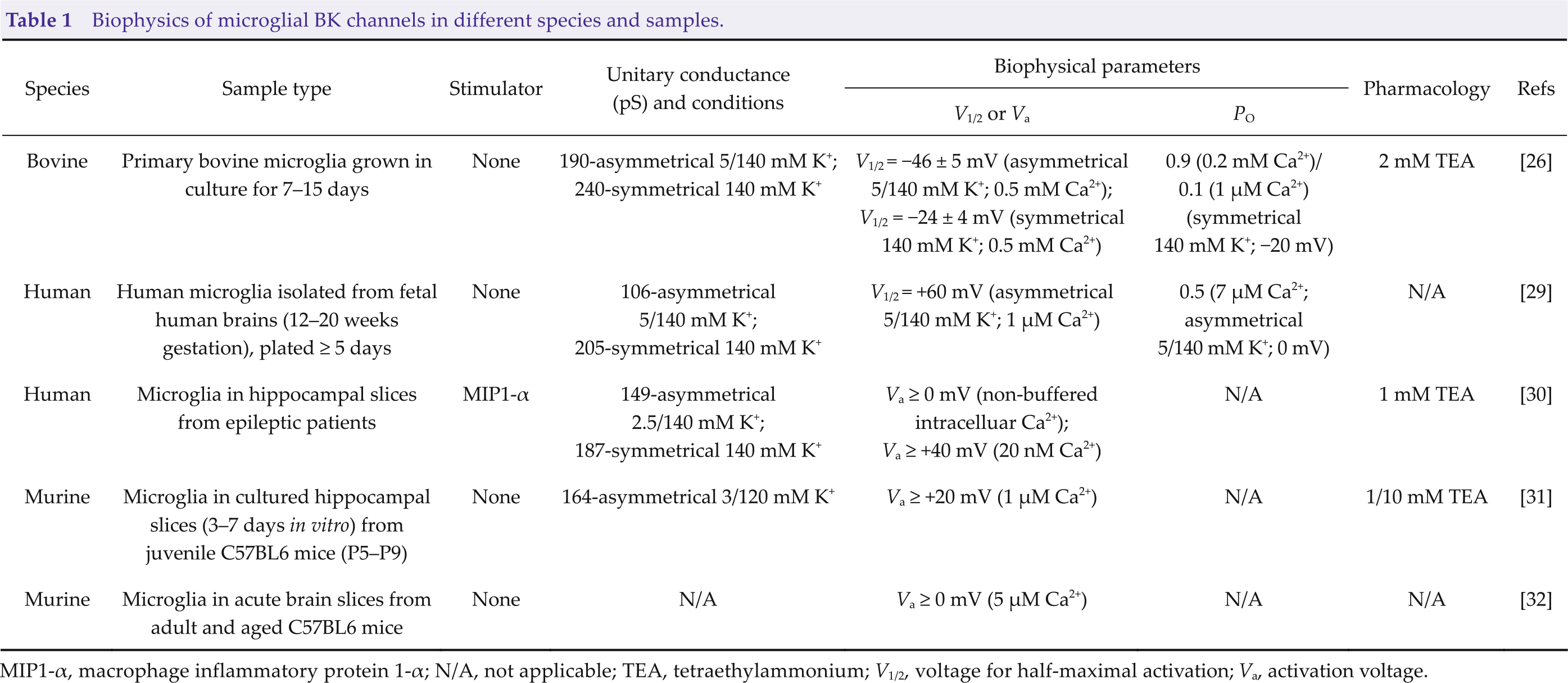
MIP1-α, macrophage inflammatory protein 1-α; N/A, not applicable; TEA, tetraethylammonium; V1/2, voltage for half-maximal activation; Va, activation voltage.
In addition, Bordey and Spencer observed increased BK currents in microglia following exposure to the chemokine MIP1-α, indicating a possible role of BK channels in microglial migration [30]. Huang et al. found that activation of BK channels by the specific activator NS19504 facilitated the phagocytosis of both fluorescent beads and neuronal debris by primary microglia after 1 h of oxygen–glucose deprivation. Similarly, in ischemic mice, NS19504 increased the number of phagocytic microglia to decrease neuronal apoptosis [43]. These effects could be reversed by paxilline, suggesting that BK channels are involved in phagocytosis by microglia.
4 Microglial BK channels and neurological diseases
In the brain, the activation of microglia results in neuroinflammation, which correlates with many neurological diseases such as neuropathic pain [44, 45] and stroke [46, 47]. Because BK channels play crucial roles in the activation of microglia, they are also probably involved in neuroinflammation-associated neurological diseases. In fact, following a blast-induced traumatic brain injury in rats, both the mRNA and protein levels of BK channels were elevated in the cortical tissue around the damage zone. The functional blocking of this channel using paxilline significantly suppressed the expression of the nucleotide-binding and oligomerization domain (NOD)-like receptor family, the pyrin domain-containing 3 (NLRP3) inflammasome, and the production of the proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 [48]. However, because these results were obtained from cortex samples without distinguishing the cell type, it is difficult to conclude that to what extent these changes arose due to the activity of microglial BK channels.
Neuropathic pain is a common and severely disabling condition caused by a pathological process in which the activation of spinal microglia plays key roles [49]. Surgical transection of the L4 nerve in mice resulted in the hyperactivation of spinal microglia, marked by an increased number of cells and a larger soma size with shorter processes, in addition to an increased expression of mature IL-1β [35]. The BK channel blocker charybdotoxin (CTX) (4.3 ng, i.t.) significantly reversed this activation of spinal microglia and inhibited the nerve injury-induced increased expression of both P2X4 receptors and brain-derived neurotrophic factor, the two markers of the initiation of neuropathic pain, in spinal microglia [50–52]. Behavioral studies demonstrated that CTX significantly increased the paw withdrawal threshold (PWT) of mice at day 2 and 3 after nerve injury. CTX could also suppress tactile allodynia caused by lysophosphatidic acid, a causative factor in the activation of microglia and subsequent tactile allodynia [53–55]. In contrast, a low dose of NS1619 (0.072 μg, i.t.) induced the activation of spinal microglia and reduction of PWT [35].
Microglial BK channels also play similar roles in morphine-induced hyperalgesia [33]. Microglial BK currents became activated after chronic morphine treatment, which are associated with reduced PWT and hot plate latency. Blockade of BK channels with either iberiotoxin (IbTX) or paxilline markedly attenuated morphine-induced hyperalgesia. Intriguingly, after exposing wild-type (WT) or Slo1-lacking primary microglia to 10 μM morphine for 24 h in the absence or presence of paxilline, and then transferring these microglial cells to mice, only the morphine-stimulated WT primary microglia induced hyperalgesia. Both the inhibition and knockout of BK channels completely abrogated the generation of hyperalgesia. Moreover, knockdown of microglia-specific KCNMB3 with small interfering RNA (siRNA) also abolishes morphine-induced hyperalgesia. These results [33–35] indicate that microglial BK channels contribute to both neuropathic pain and morphine-induced hyperalgesia.
In a transient middle cerebral artery occlusion (tMCAO) experimental stroke mouse model [43], BK channels were found to be primarily expressed in microglia and their mRNA and protein levels in the peri-infarct area of mouse brain were reduced after tMCAO. The specific BK channel opener NS19504 reduced neuronal apoptosis, increased the survival rate, and ameliorated neurological deficit and motor function through enhanced phagocytosis by microglial cells after ischemic stroke. Blockade of BK channels with paxilline resulted in the opposite effect. These results suggest that microglial BK channels are a potential target for developing ischemic stroke therapies.
Although the abovementioned studies were conducted using systemic administration of specific channel openers/blockers or global genetic deletion of the genes encoding BK channels, it is certain that the functions of microglial BK channels are closely related to neuropathic pain, morphine-induced hyperalgesia, and tMCAO- induced ischemic stroke. Future research exclusively manipulating the microglial BK channels is needed to better clarify the role and underlying mechanism of this channel in the abovementioned neurological diseases.
5 Concluding remarks
In summary, microglial BK channels possess a large unitary conductance of 106–240 pS under different measurement conditions, prominent calcium and voltage dependence, and good sensitivity to various BK channel modulators, including TEA, CTX, IbTX, paxilline, and NS1619. These channels regulate the activation of, phagocytosis by, and probably the migration of microglia. Altered expression and function of these channels in microglia are associated with various neurological diseases such as neuropathic pain, morphine-induced hyperalgesia, and stroke.
To date, the mechanisms underlying the functions of BK channels in microglia are still elusive. Answers to the following questions would provide further insights into the regulation and function of microglial BK channels and could be beneficial for the development of therapies for neurological diseases related to the dysfunction of microglial BK channels.
1) Which types of accessory subunits are expressed in microglia? In one study, single-cell polymerase chain reaction analysis revealed that subunit β3 rather than subunit β1, β2, or β4 is exclusively expressed in spinal microglia [33]. However, because the authors of that study did not assay the γ and Lingo subunits, the expression of these auxiliary subunits in microglia cannot be ruled out. Whether microglial cells from different brain areas contain different accessory subunits still needs to be clarified.
2) Are the microglial BK channel subunits encoded by a gene with splice variants, or do they differ due to certain posttranslational modifications? If it is due to splice variants, efforts could be made to find splice variant-specific drugs to modulate BK channel activity exclusively in microglia.
3) What is the functional role and composition of the nuclear BK channels in microglia? We disclosed the presence of BK channels on microglial nuclear membranes and found that these channels are involved in the LPS-induced activation of microglia [12]. However, the mechanism underlying their function is not completely understood. Membrane-permeable and -impermeable channel blockers and electrophysiological recordings in isolated nuclei might be helpful for addressing this problem.
Finally, microglia-specific research tools such as transgenic animals with microglial BK channel knockdown or microglia-targeted drug delivery systems are urgently required to enable further investigation in this area.
Footnotes
Conflict of interests
The contributing author reports no conflict of interests in this work.
Funding
This work is granted by the National Natural Science Foundation of China (Grant No. 31400924) and the Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD).